DOI: 10.31038/PSC.2021111
Abstract
Background: Juvenile hyaline fibromatosis (JAF) is a rare autosomal-recessive disease in which patients progressively develop cutaneous tumoral fibroblastic proliferations, and joint contractures with bone involvement. JAF is caused by aberrant synthesis of glycosaminoglycans by fibroblasts due to a mutation of the capillary morphogenesis factor-2 gene (CMG2). Limited treatment options are available.
Method: We report monozygotic twins who presented with multiple, recurrent, painless cutaneous nodules.
Result: The presence of twins with JAF is extremely rare. A lesion on the head of one boy had ruptured, and pathological analysis indicated benign spindle cells in a periodic acid-Schiff (PAS)-positive hyaline background. One of twins had much more severe clinical presentation than the other, including more frequent diarrhea, larger nodules, more severe joint involvement, and more easily ruptured masses.
Conclusion: Monozygotic twins who present with JAF may have different severity of symptoms despite the presence of identical mutations in CMG2.
Keywords
juvenile hyaline fibromatosis, twins, subcutaneous mass
Introduction
Juvenile hyaline fibromatosis (JHF) is a rare autosomal-recessive disease caused by mutations in capillary morphogenesis gene-2 (CMG2). There have been fewer than 70 cases reported so far in the literature [1–2], and there has only been one report of twins with JHF [3].The clinical onset usually occurs before 5 years of age, and this disorder is slightly more common in boys [4]. JHF is characterized by cutaneous tumoral fibroblastic proliferation, joint contractures, and bone involvement [2].
Materials and Methods
Clinical Features
Two one-and-a-half year-old male monozygotic twins presented with multiple, progressive, painless, variable-sized nodules all over their bodies. The parents had the same family name and lived in the same remote village in Henan province of China, but were not first-degree relatives. In the year prior to presentation, both infants had recurrent diarrhea of unknown etiology, and in the 6 months prior to presentation, both had multiple, painless, variable-sized cutaneous nodules all over their bodies. These nodules were first noticed around the joints, then on the trunk and head, and eventually all over their bodies. The size of nodules increased gradually over time. One year ago, a nodule on one child’s finger was given an incisional biopsy at a local hospital, and the pathological result was “benign”, but there was no definite diagnosis or further investigation. When they were hospitalized, one of masses on the head had decayed spontaneously and the joint contractures had deteriorated gradually, which crippled both of them. They both had very poor appetites. One of them (patient A) had more severe signs and symptoms than the other (patient B) [Table 1]. They both achieved normal mental development milestones.
Table 1. Clinical feature difference between the twins
|
|
patien A |
patient B |
|
clinical onset |
earlier |
later |
|
maximium diameter of nodules |
25 |
15 |
|
mass rupture |
yes |
no |
|
gingival proliferation |
svere |
moderate |
|
joint constracture |
svere |
moderate |
|
perianal tissue proliferation |
yes |
no |
|
nostril narrow |
yes |
no |
|
diarrhea |
conatantly |
intermittent |
On physical examination, there were multiple, painless, variable-sized nodules all over the bodies, which were soft upon palpation [Figure 1]. The largest nodule was on the head of patient A, whose diameter was 25 cm [Figure 2]. One mass on the head had ruptured and had a terrible odor [Figure 3]. Cutaneous tumoral fibroblastic proliferation around the lips and gingival hyperplasia [Figure 4] were also present. Patient A had a narrow nostril caused by subcutaneous proliferation, but no shortness of breath [Figure 5]. There were many nodules on their hands and fingers [Figure 6], elbows [Figure 7], and abdomens [Figure 8]. There were also flexion contracture deformities of the limbs [Figure 9], scales on the feet [Figure 10], and proliferation in the perianal area [Figure 11], but no significant lymphadenopathy or hepatosplenomegaly.
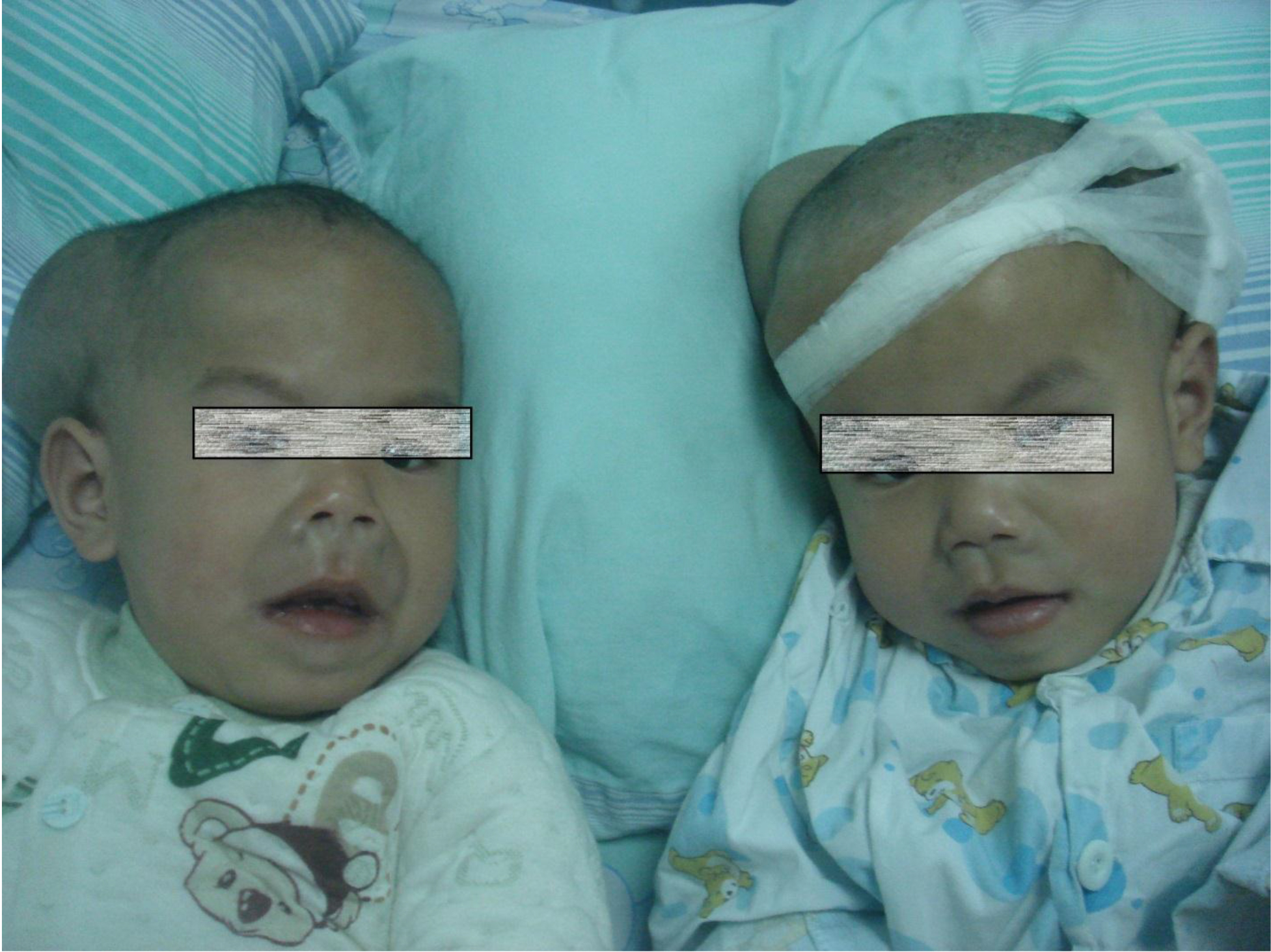
Figure 1. Appearance of the twins.
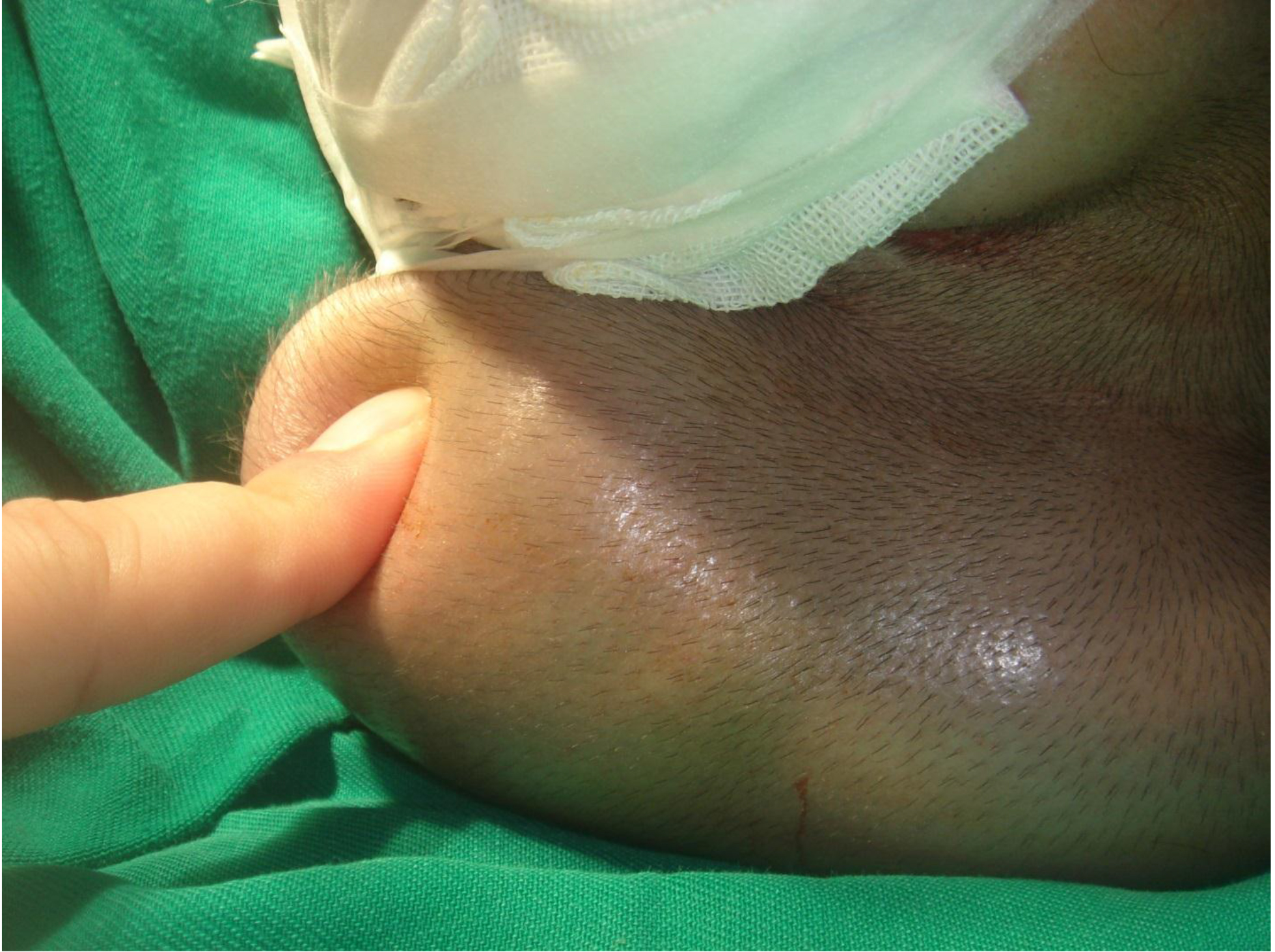
Figure 2. Mass on the head.
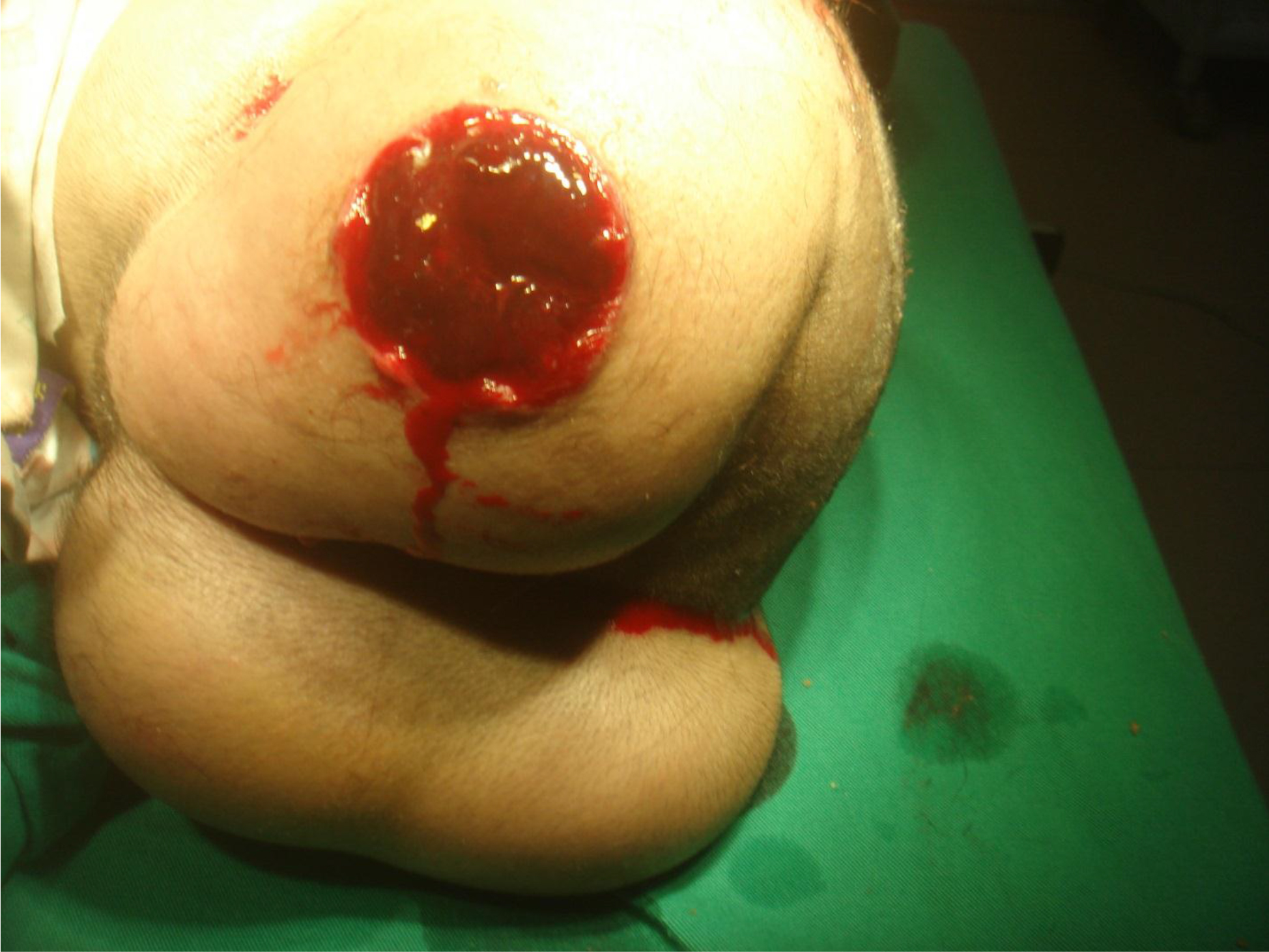
Figure 3. Decayed mass on the head.
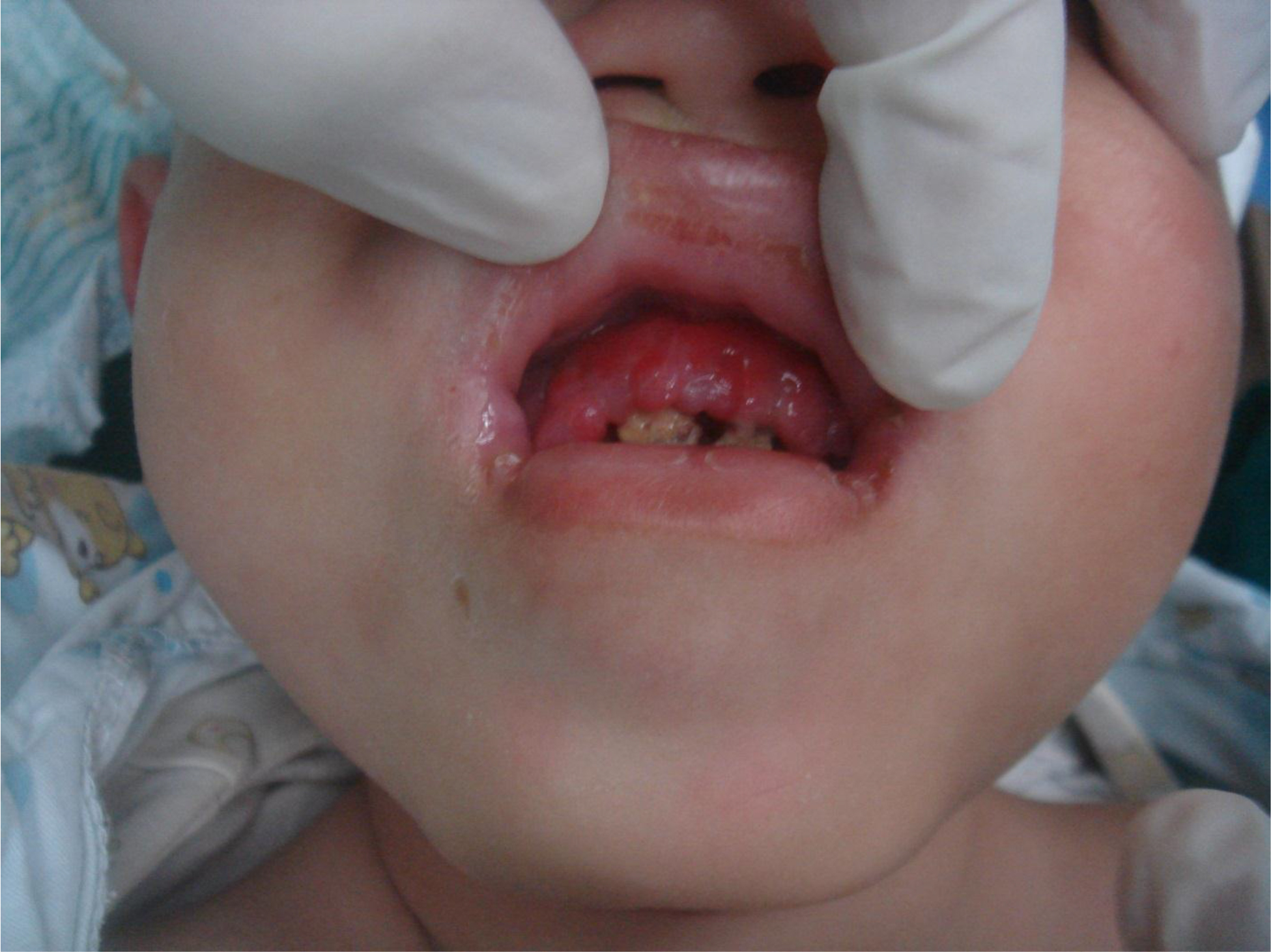
Figure 4. Cutaneous proliferation around the lips and gingival hyperplasia.
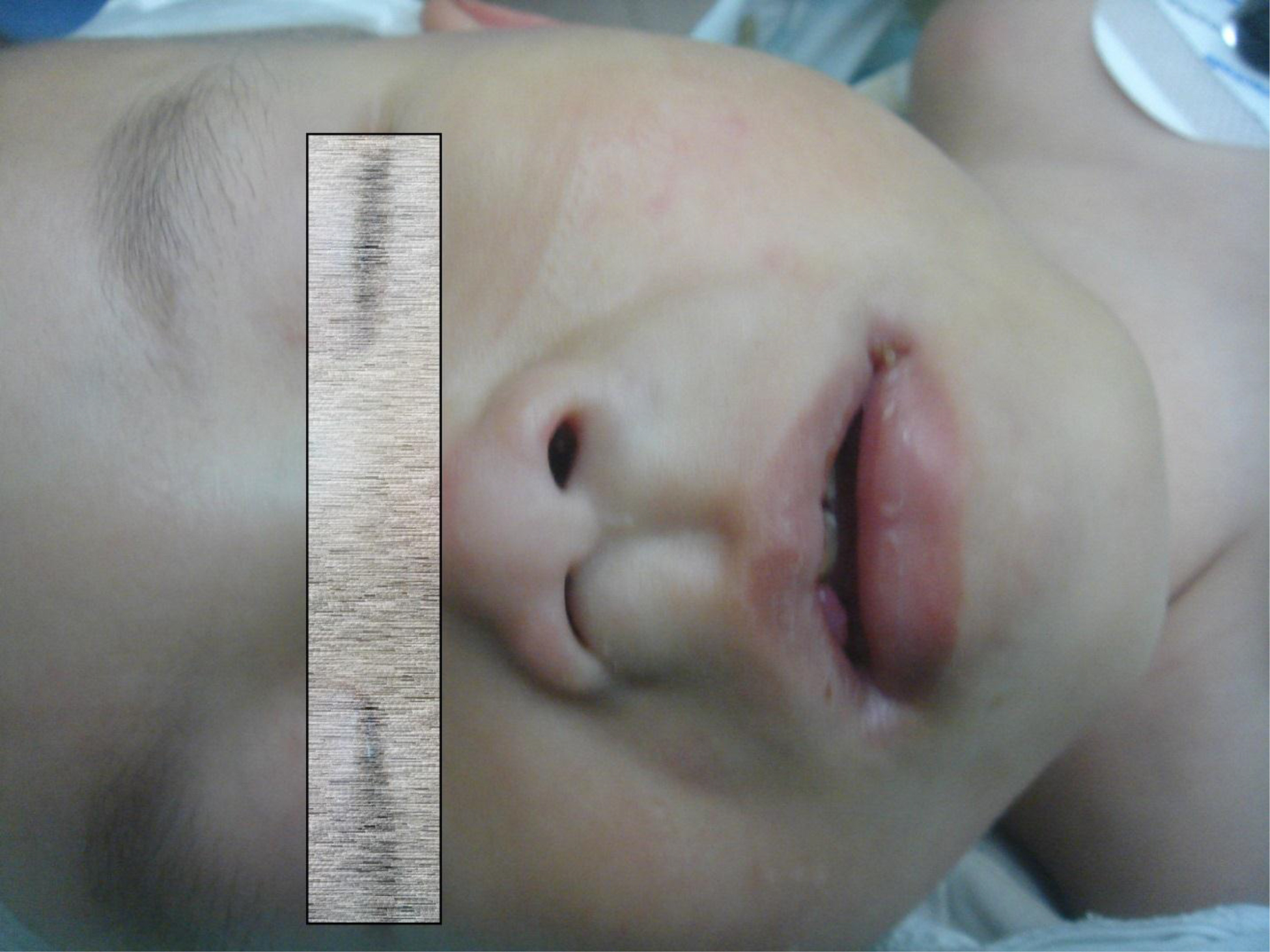
Figure 5. Patient A had a narrow nostril.
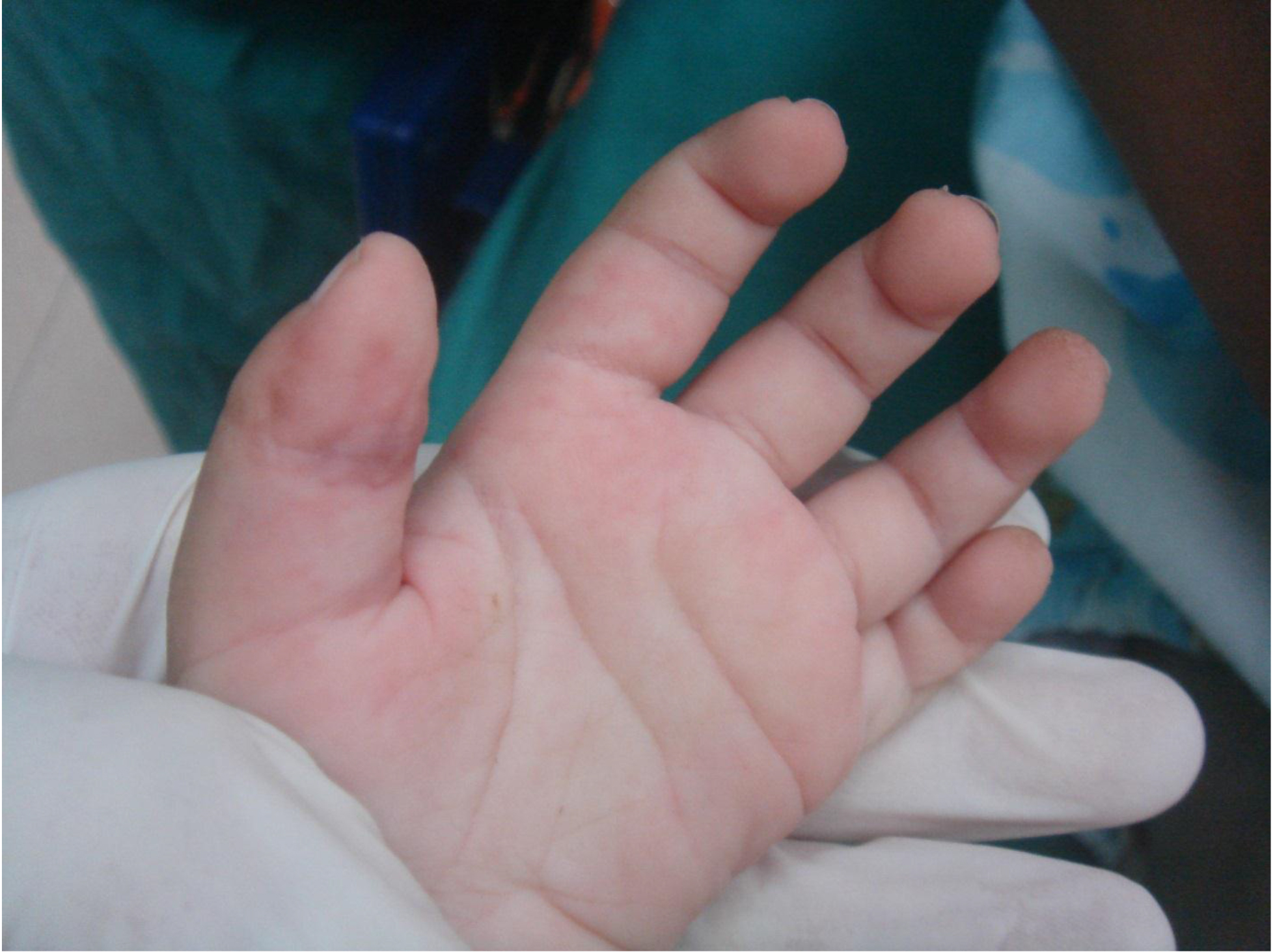
Figure 6. Nodules on hands and fingers
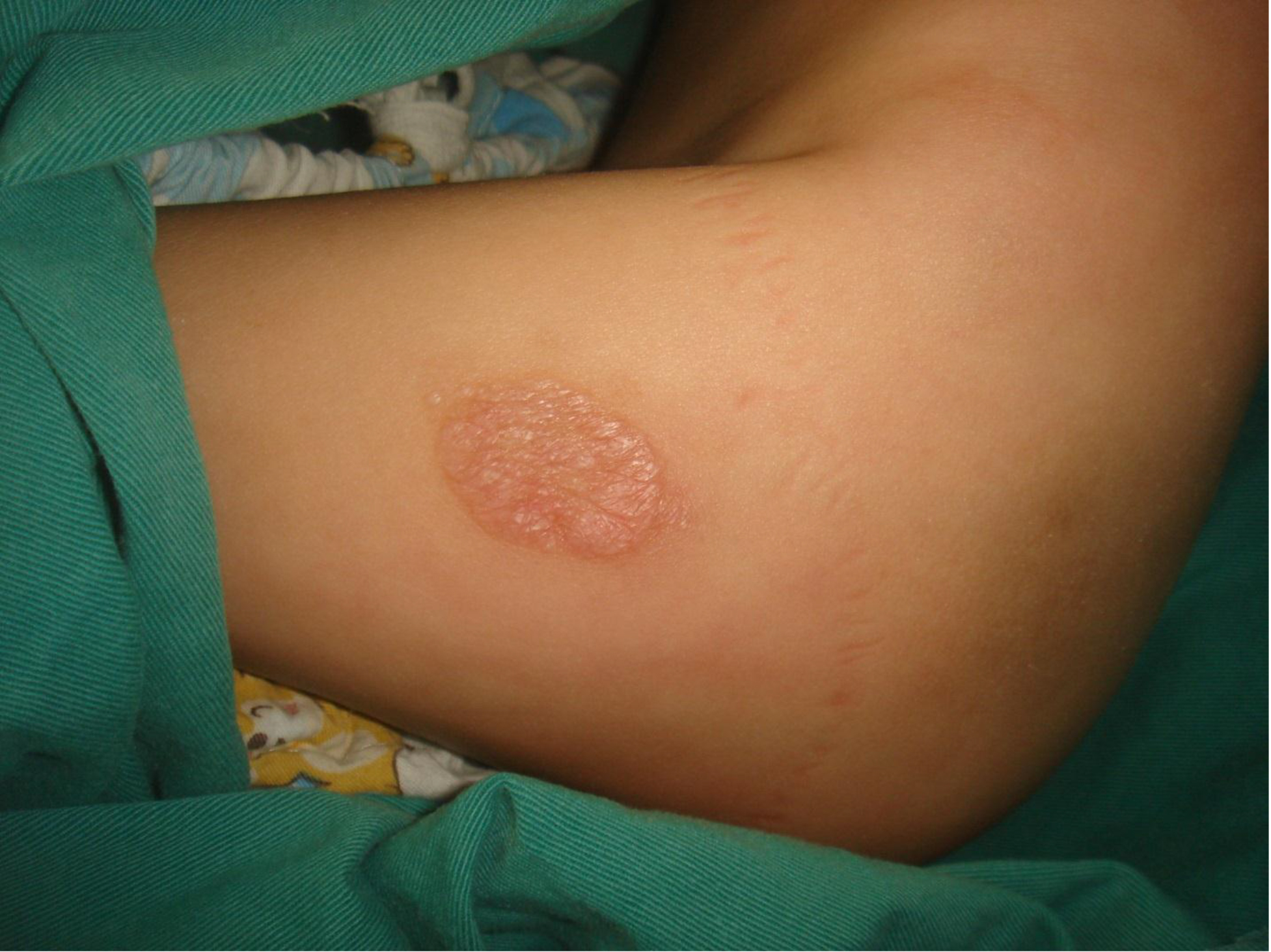
Figure 7. Nodules on elbow.
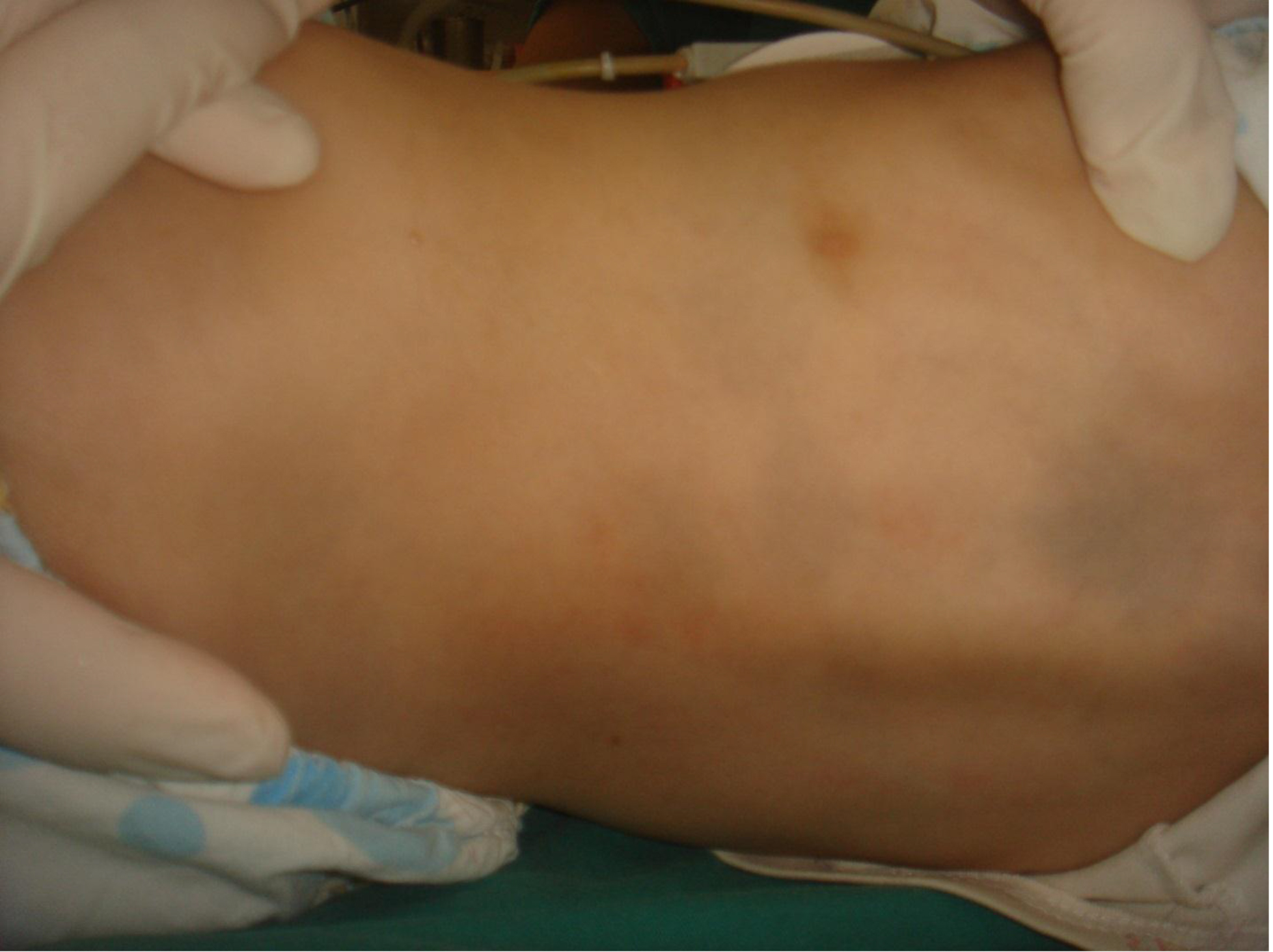
Figure 8. Nodules on abdomen.
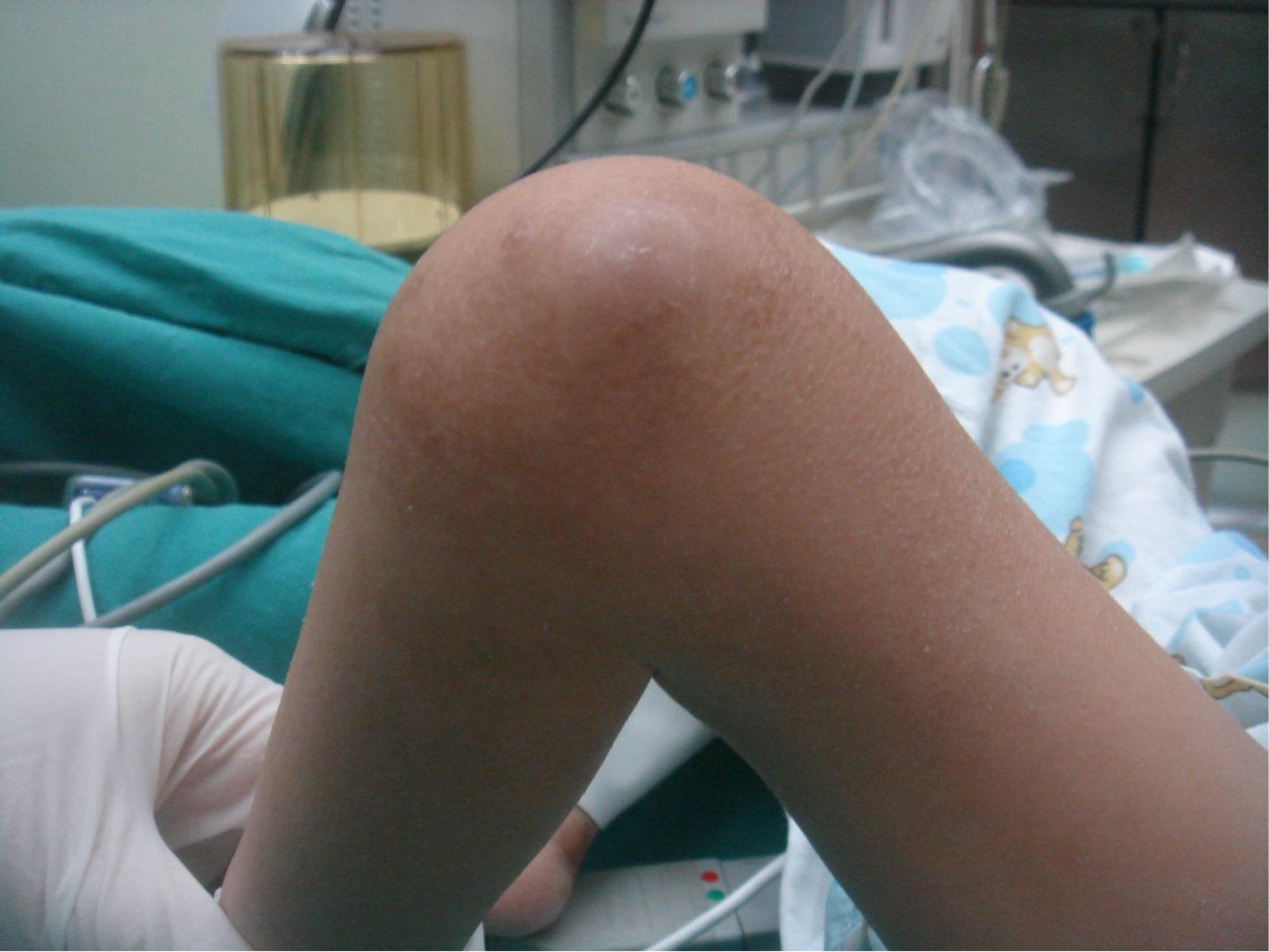
Figure 9. Flexion contracture deformities of the knee.
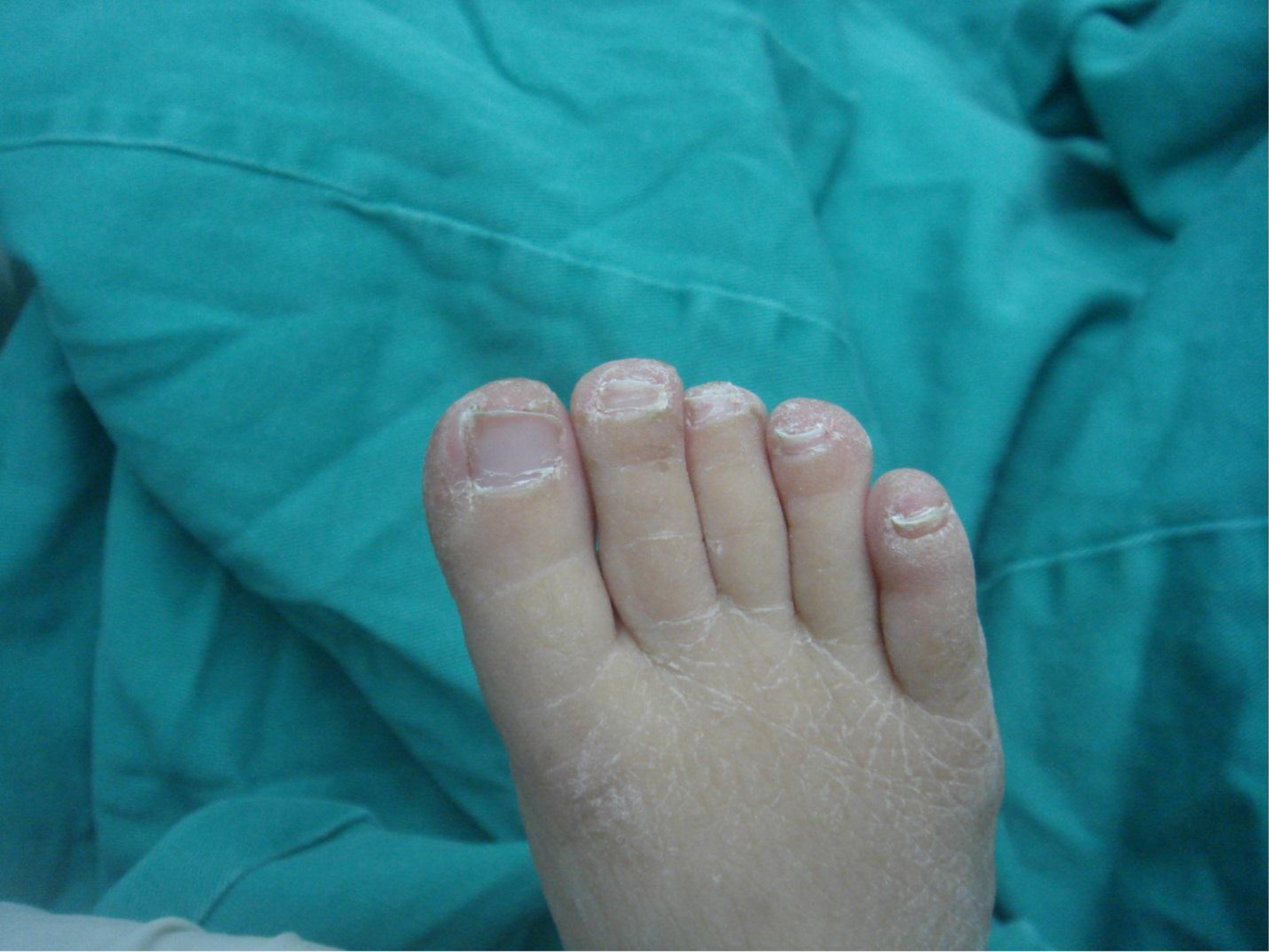
Figure 10. Scales on the foot.
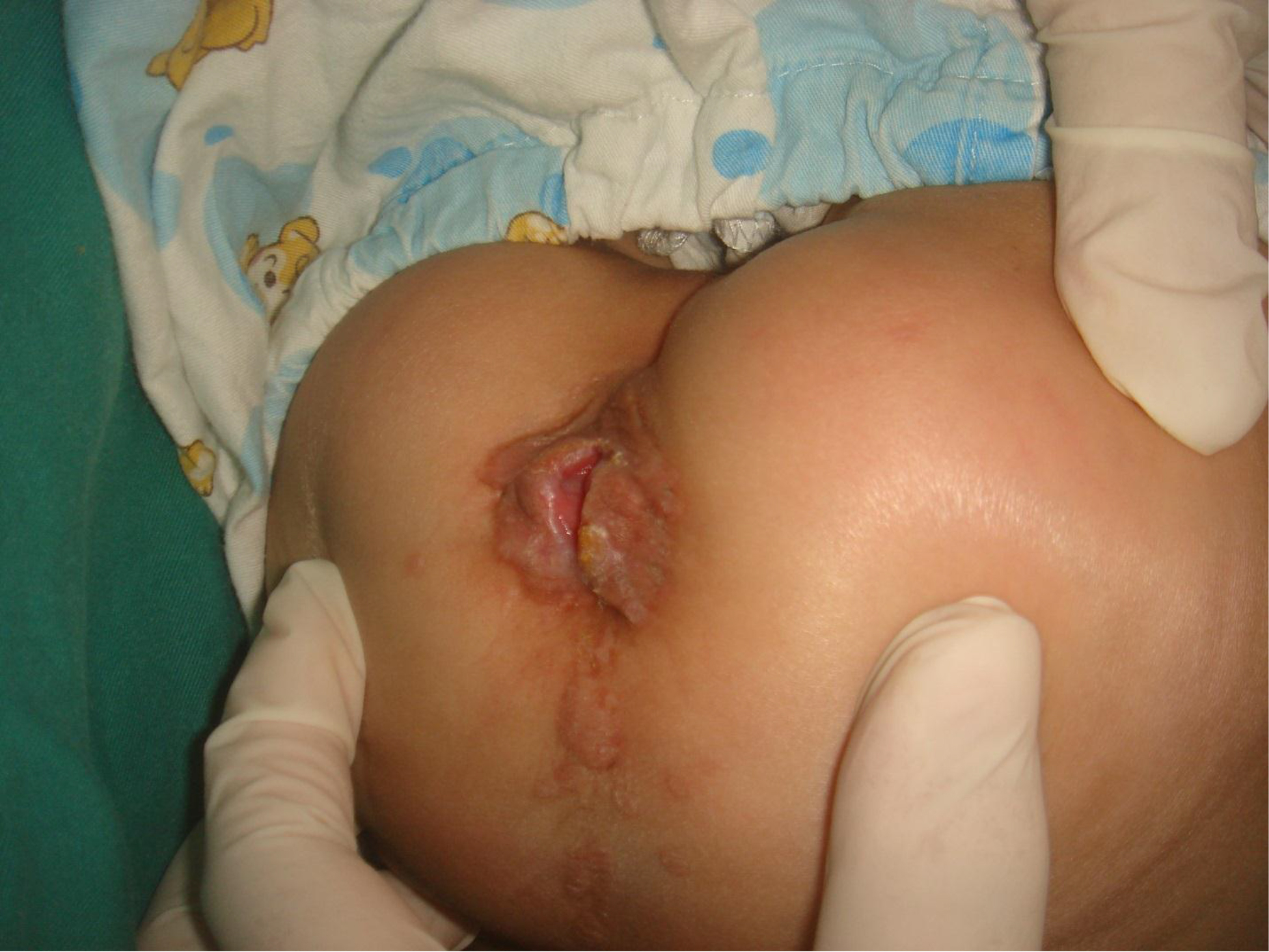
Figure 11. Proliferation in the perianal area.
The complete blood counts and electrolyte levels were within normal limits. An X-ray indicated osteoporosis in vertebrae of patient A [Figure 12]. Computed tomography (CT) results showed no masses in the abdominal cavities, except for cutaneous nodules in the abdominal wall [Figure 13]. A head CT of patient A showed a large subcutaneous mass but no intracranial abnormalities [Figure 14].
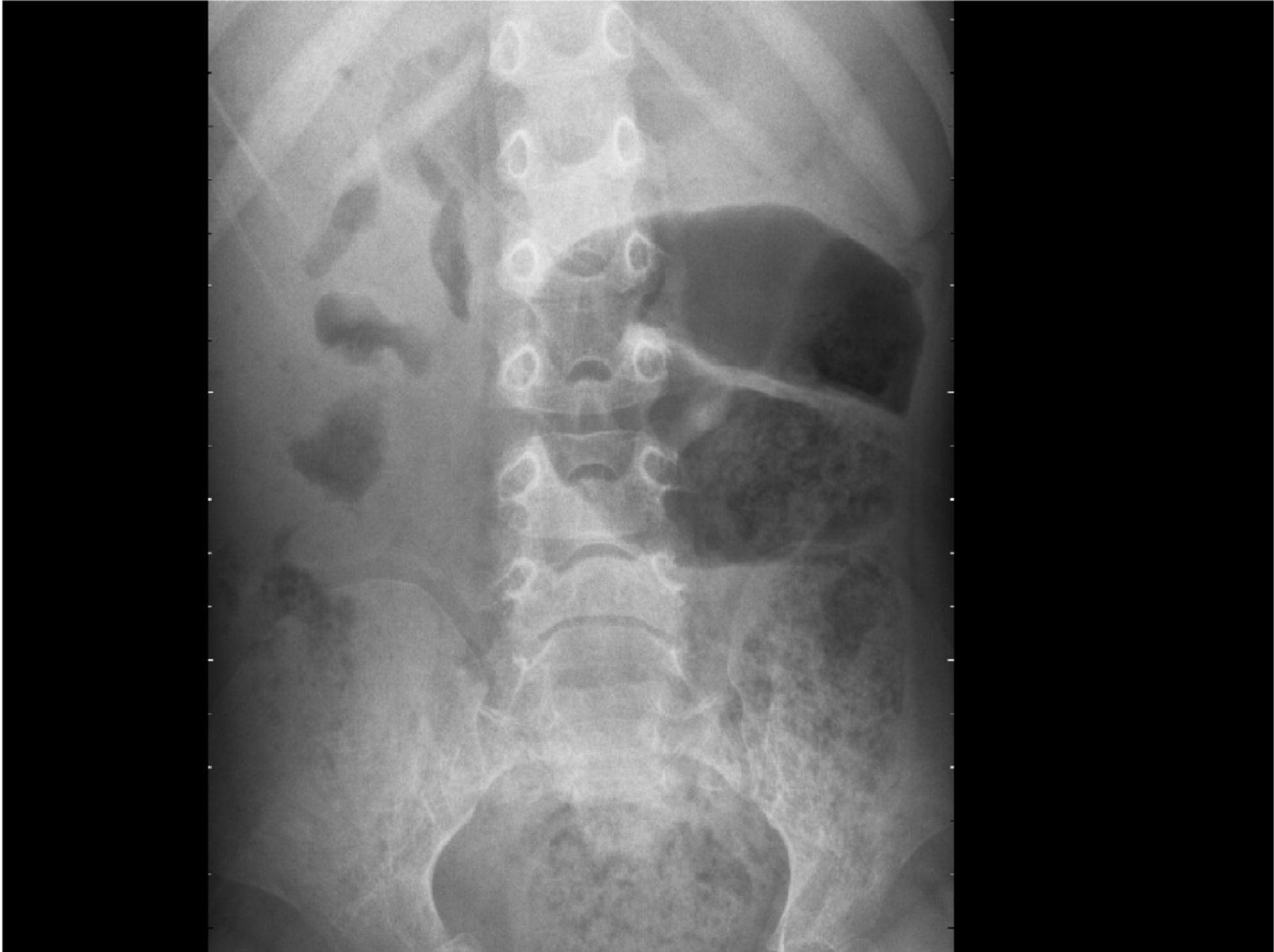
Figure 12. Osteoporosis in vertebrae.
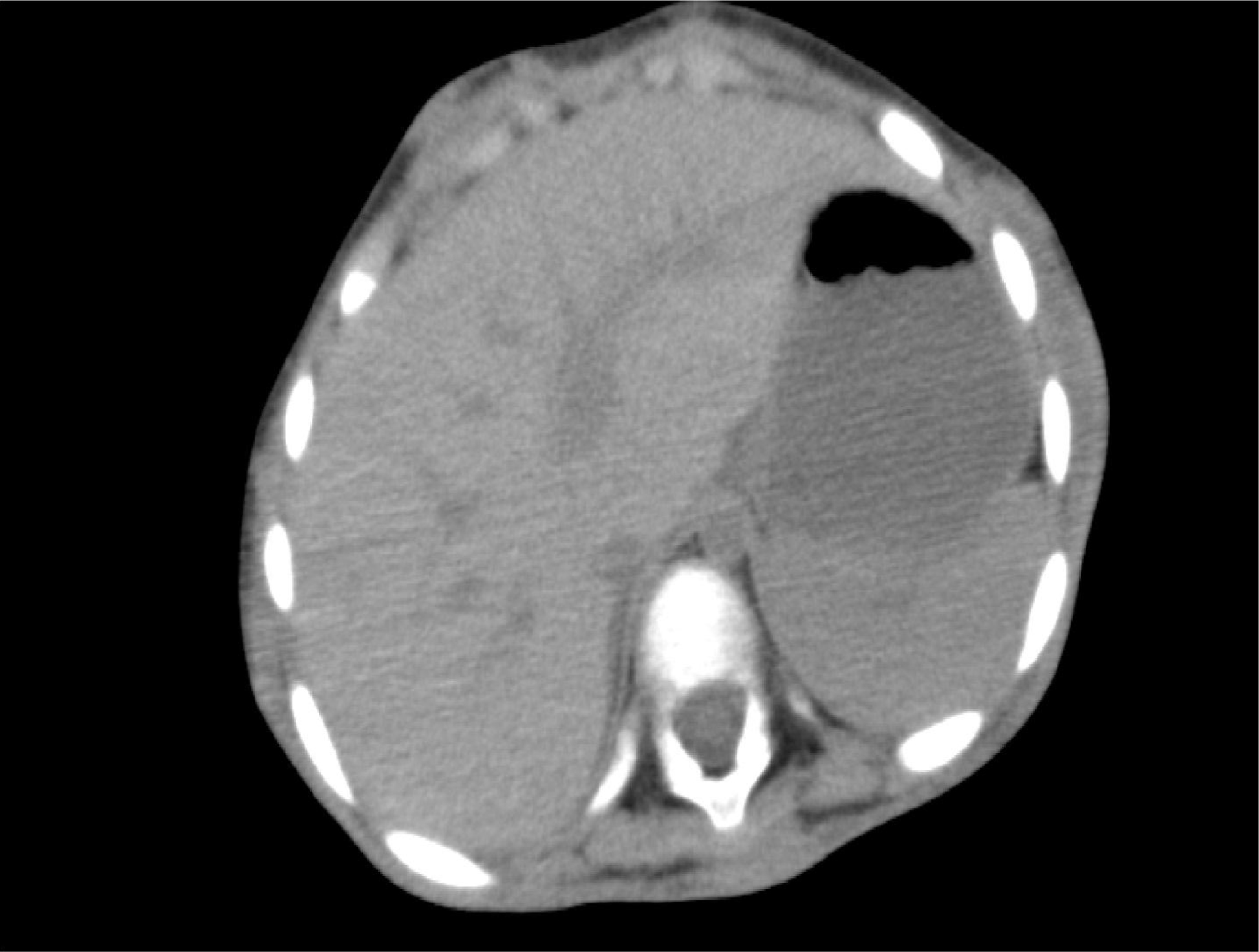
Figure 13. Cutaneous nodules in the abdominal wall.
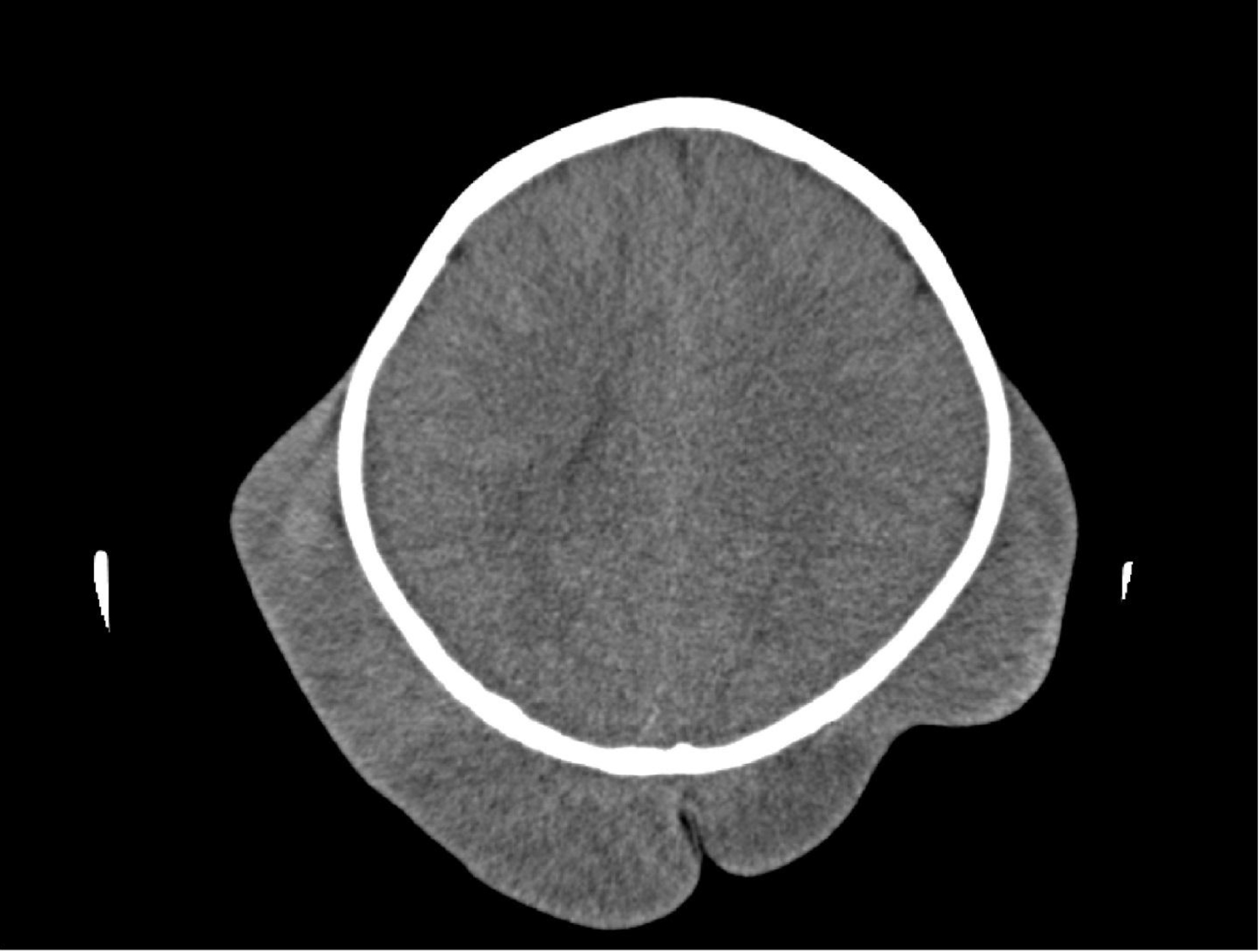
Figure 14. Subcutaneous mass without intracranial abnormalities.
After consent from the parents, a biopsy of the large mass on patient A’s head was performed. Hyaline subcutaneous tissue was present beneath the skin [Figure 15], which was sent for pathological investigation. The ruptured head mass was covered with dressing and required regular changes.
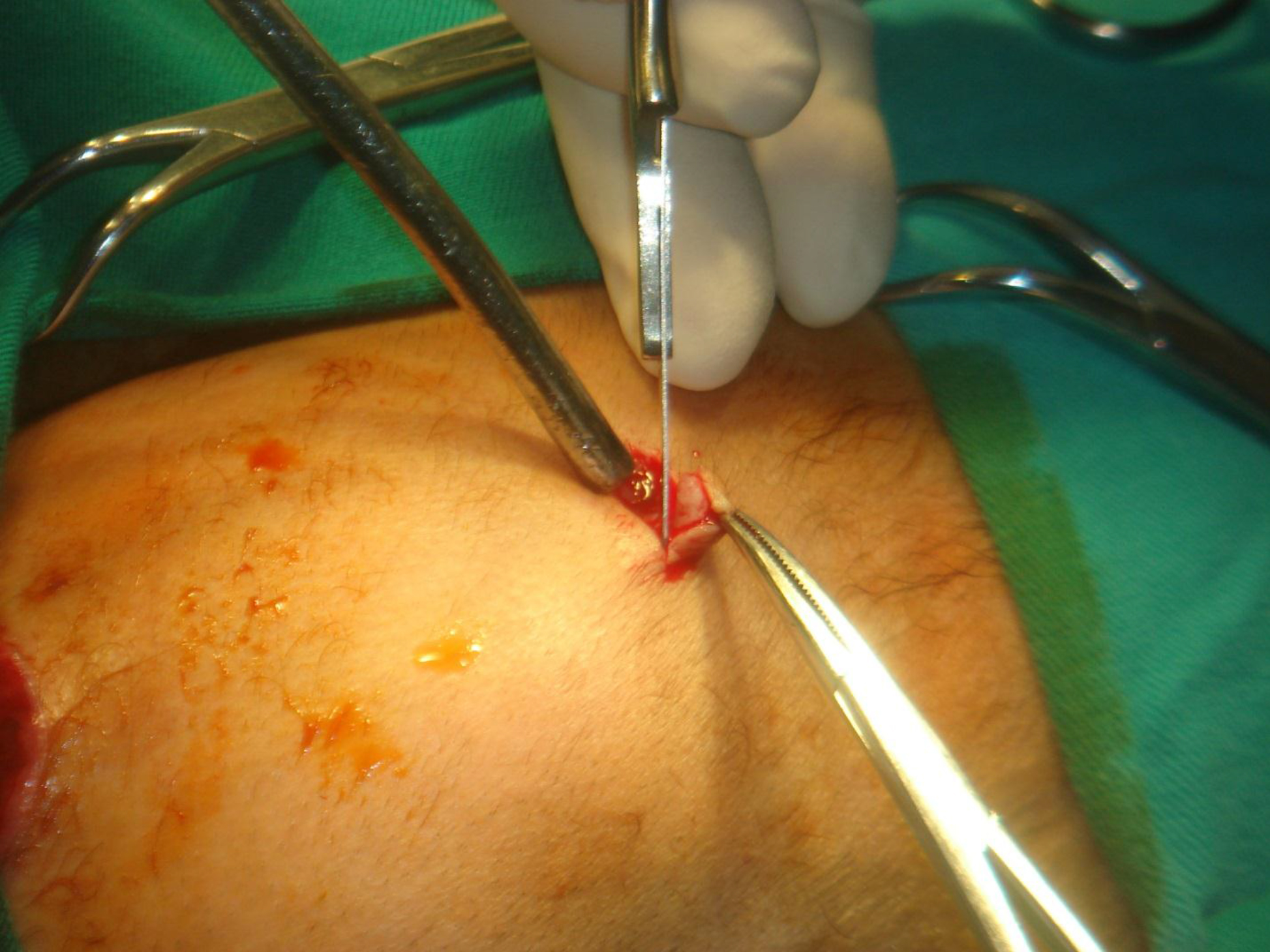
Figure 15. Hyaline subcutaneous tissue beneath the skin.
Pathological Findings
Gross examination indicated that the nodules had a gelatinous white and hyaline appearance [Figure 16]. Microscopically, there were poorly circumscribed lesions composed of many uniform spindle cells embedded in an abundant homogenous fibrous matrix. This matrix was eosinophilic and tested positive on periodic acid-Schiff (PAS) staining, findings characteristic of JHF [Figure 17]. The parents did not consent to surgical procedures on the other child.
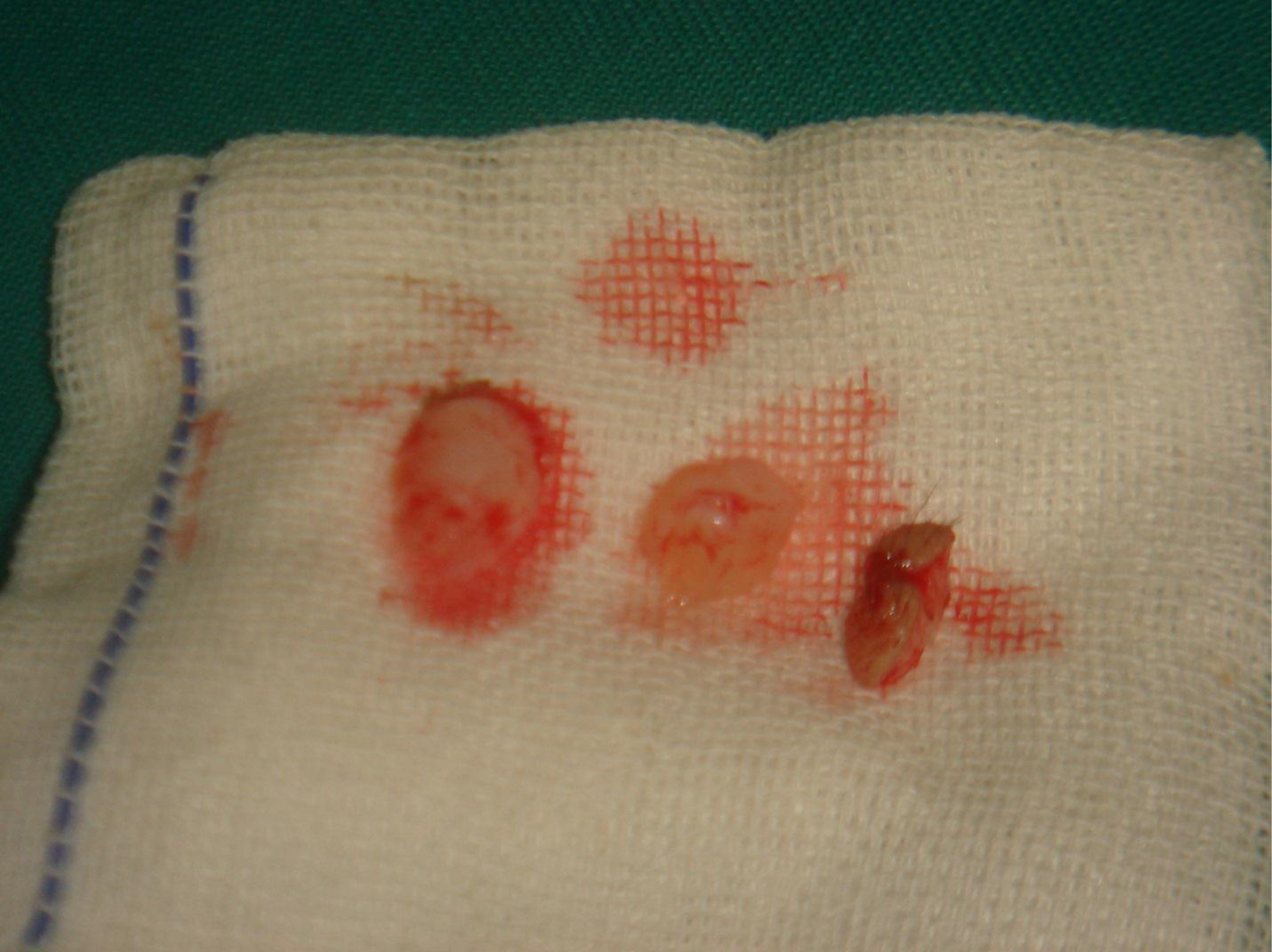
Figure 16. Gross pathological examination.
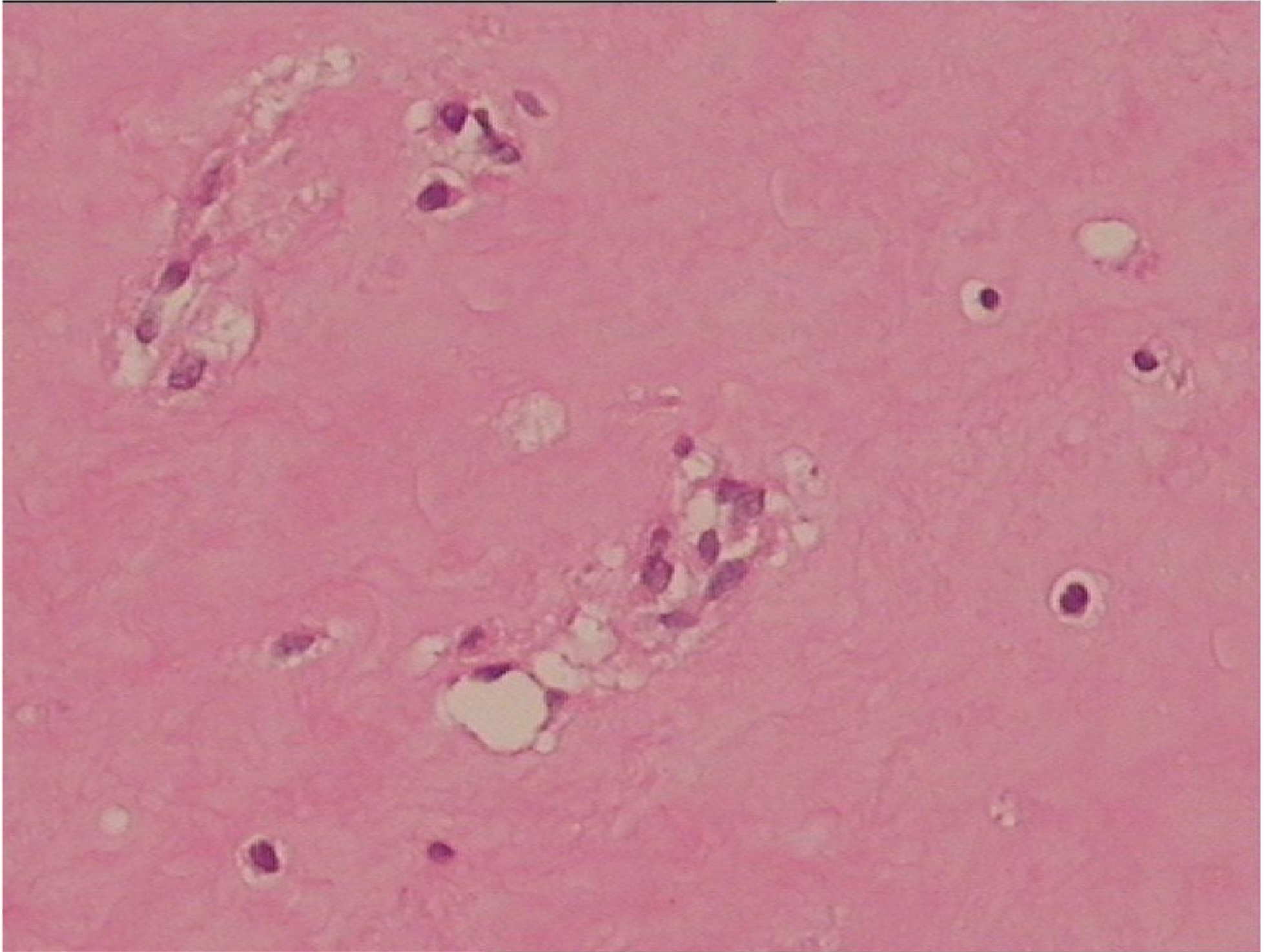
Figure 17. Microscopical pathological examination.
RNA isolation and cDNA amplification of samples from each patients’ blood were performed. Cloning and sequencing of the CMG2 gene showed that both patients had the same 4 mutations: c.294C→A, c.1069G→C, 1074delT, and c.1153G→C.
Follow up
The twins were followed up for 4 years after the initial presentation. At the last follow-up, the decayed head lesion of patient A was still unhealed, there were more nodules all over their bodies, and the masses on the head of patient A were larger. The parents refused further therapy.
Discussion
JHF is a rare autosomal-recessive disease that was first described by McMurray [1] as molluscum fibrosum, but renamed by Kitano [2]. Patients with JHF present with multiple cutaneous nodules or masses, gingival hypertrophy, joint contractures, and osteolytic lesions [4]. Skin lesions are the most prominent symptoms [8]. Normal motor development is impaired if joint contractures occur during infancy. Eating and delayed dentition can be caused by severe gingival hyperplasia [1].
Histological examination can confirm a diagnosis of JHF [10]. In particular, lesions from the dermis, subcutis, gingivae, bone, and joints contain abundant homogeneous eosinophilic matrix, which is embedded with cords of spindle-shaped cells. The matrix stains positively with PAS and alcian blue, but not with toluideine blue or Congo red [10]. Nodules are calcified occasionally, but have no elastic tissue. The etiology is unknown. The differential diagnosis includes neurofibromatosis, fibromatosis, amyloidosis, infantile systemic hyalinosis, lipoid proteinosis, and Winchester syndrome [2].
The JHF gene is located on gene 4q21 [5], and previous studies have described the effects of mutations in CMG2 [6]. Previous researchers have described JHF as a connective tissue disease that is characterized by aberrant synthesis of glycosaminoglycans. Dermatan sulfate is the predominant glycosaminoglycan in the skin of patients with JHF, which also has chondroitin sulfate and hyaluronan, but hyaluronan is the most abundant glycosaminoglycan in normal skin [7].
There are no specific treatments for JHF, but cosmetic surgery and those that limit orthopedic disability may be employed. This disease has a progressive course, and most patients only survive up to the 4th decade [1–2]. Relapses are common after tumor removal. A previous report indicated good cosmetic results after removal of more than 100 tumors over a period of 19 years [9]. Physiotherapy may be performed to prevent flexion contractures.
The occurrence of twins with JHF is extremely rare, and there has only been one previous case report [3], and the researchers did not report differences between these twins. It is generally accepted that twins will have the same presentations, because JHF is a genetic disease. However, our twins had very different presentations. Patient A had more severe symptoms [Table 1], and a head mass that ruptured and bled, and was refractory to treatment. Patient A also had more frequent diarrhea, larger nodules, and more severe joint involvements.
We do not know the reasons for the different presentations of these twins. They had identical mutations in CMG2, and might be expected to eventually develop the same signs and symptoms. It may be hypothesized that the differences between the normal and genetically mutated somatic cells led to the differences in these twins. It is possible that patient B will eventually develop the same severe symptoms as patient A. A close follow-up of these children is warranted.
Statement
There are no prior publications or submissions with any overlapping information, including studies and patients. The manuscript has not been and will not be submitted to any other journal. There have no financial support or relationships that may pose conflict of interest. Gaoyan, Deng wrote the first draft of the manuscript. No honorarium, grant, or other form of payment was given to anyone to produce the manuscript. Each author listed on the manuscript has seen and approved the submission of this version of the manuscript and takes full responsibility for the manuscript.
References
- Nischal KC, Sachdev D, Kharkar V, Mahajan S. (2004) Juvenile hyaline fibromatosis. J Postgrad Med 50: 125–6.
- Haleem A, Al-Hindi HN, Juboury MA, Husseini HA, Ajlan AA. (2002) Juvenile hyaline fibromatosis: Morphologic, immuno histochemical and ultrastructural study of three siblings. Am J Dermatopathol 24: 218–24.
- Habibeddine S, Khadir K, Azzouzi S, Skalli S, Lakhdar H. (2003) Juvenile hyaline fibromatosis: 2 twin brothers affected. Ann Dermatol Venereol 130: 43–6.
- Weiss SW, Goldblum JR. (2001) Fibrous tumors of infancy and childhood. In: Strauss M, editor. Enzinger and Weiss’s Soft Tissue Tumors. 4th ed. St. Louis: Missouri Mosby; 363–7.
- Nazneen Rahman, Melanie Dunstan M. Dawn Teare. (2002) The gene for juvenile hyaline fibromatosis maps to chromosome 4q21. Am J Hum Genet 71; 975–980.
- Sandra Hanks, Sarah Adams, Jenny Douglas. (2003) Mutations in the gene encoding capillary morphogenesis protein 2 cause juvenile hyaline fibromatosis and infantile systemic hyalinosis. Am J Hum Genet 73; 791–800.
- Katagiri K, Takasaki S, Fujiwara S, Kayashima K, Ono T, et al. (1996) Purification and structural analysis of extracellular matrix of a skin tumor from a patient with juvenile hyaline fibromatosis. J Dermatol Sci 13: 37–48.
- Sanzalin H, Kiyozuka Y, Uemura Y, Shikata N, Ueda S, et al. (1998) Juvenile hyaline fibromatosis: A report of two unrelated adult sibling cases and a literature review. Pathol Int 48: 230–6.
- Woyke S, Domagala W, Markiewicz D. (1984) A 19-year follow-up of multiple juvenile fibromatosis. J Pediatr Surg 19: 302.
- Arnold H, Odom RB, James WD. (1990) Andrews’ diseases of the skin, Clinical dermatology. 8th ed. Philadelphia: Saunders, 712.