DOI: 10.31038/JMG.2019215
Summary
The modular folding patterns are different from 5’UTR, coding region and 3’ UTR. The 5’ UTR folds into more stable form per unit chain length than coding region and 3’ UTR. These differences appear to be universal for many mature mRNAs. The 3’ UTR folding is least stable where the miRNA’s target preferentially. The local screening of the splice region between exon 1 and exon 2 reveals a stable hairpin structure which may have regulatory role in splicing. The mutation sequence in BL22 lymphoma introduced poly C sequences and regional folding produces in favor of spliceosome formation than germ line sequences. The changes may become a drug targetable site, because the mutation into –CCCCC- located near the stable hairpin and it can be cancer specific target to inhibit regulatory hairpin at the splice site.
Introduction
The human Myc gene is located at the chromosome 8q24 and expressed throughout the cell cycle and mitogen stimulation (increase Myc protein) or terminal differentiation (decrease Myc protein) leads to different levels of Myc gene expression and the protein levels. The c-myc gene has four promoter sites designated as P0, P1, P2 and P3 (Figure 1). The P3 is located near 3’ end of intron 1. They are independently regulated transcription initiation sites. In HL60 cells, the expression of c–myc RNA from these promoters are 5% (P0), 10.25% (P1) and 75–90% (P2). The P3 is responsible for ~5% of total c-myc mRNA. The sizes of mature mRNAs from these promoters are 2.5–3.1 kb. 2,2 kb, 2.4 kb and 2.3 kb for the P0, P1, P2 and P3 respectively [1]. The protein of 114 amino acids called mrtl (myc-related transcription/localization regulatory factor) is produced from transcript nucleotide 1,731–2,075 (Accession X00364) between P0 and P1. This protein is transmembrane protein distributed at the nuclear membrane and contiguous to endoplasmic/nucleoplasmic reticulum. It binds to translation initiation factor eIF2α, the 40S ribosomal protein RACK1 and the c-myc mRNA [2]. The c-myc gene contains pause /block/termination sites at +30 nucleotides (proximal to TSS2) and at +371/+421 nucleotides (end of exon 1 and beginning of intron 1) from P2 promoter [3] [4] [5]. It also contains two poly(A) sites at the end of the gene. In the experiments with human c-myc gene construct and HeLa cell extracts, it was found that the majority of transcription is from P2 and RNA polymerase II and III can initiate transcriptions. However, the pol III transcription terminates at the end of exon 1 where elongation block sequence motifs are present for pause or termination. The pol II transcription is also paused at the exon1 and intron 1 junction but reads through depending on the context of translocation, mutation, mitogen treatment or retinoic acid treatment [5]. In case of disruption in intron 1 during translocation (B-cell lymphoma Manca), the cryptic promoter within intron 1 or close to translocated c-myc gene under the influence of the immunoglobulin heavy chain enhancer, enables transcription of exon 2 and exon 3 from the translocated c-myc genes. In HL60 promyelocytic leukemia cell line, the exon 1 transcription over exon 2 transcription is ~3 fold in excess while when cells treated with retinoic acid, the ratio increases up to 15 folds. The retinoic acid treatment of HL60 cells induces differentiation into granulocytes and overall c-myc gene expressions reduce more than 10 fold in a coordinated manner from P0, P1 and P2 promoters at 21 hours and at 6 days when differentiation is complete, the expression from P0 and P1 are not detectable and only P2 expressions are detectable. The runoff transcription of c-myc gene in HL60 nuclear extract also reveal the bidirectional transcription upstream of exon 1 utilizing P0 and P1 promoters [6]. The DMSO (dimethylsulfoxide) treatment, a potent inducer of granulocyte differentiation in HL60 cells, also causes rapid decrease of cytoplasmic c-myc RNA by elongation block within untranslated c-myc leader sequence [4].
Figure 1. The c-Myc Gene Map

The c-myc gene contains three exons in ~5.2 kb transcriptional unit. There are two transcription initiation sites in exon 1 which are 160 bp apart [7]. The start site 1 expresses ~555 nt exon 1. The TSS 3 in intron 1 is also identified. Two translation initiation sites are present in mature c-myc mRNA , one at the end of exon 1 (CUG initiation codon) and the other one at the exon 2 (AUG initiation codon) producing 67 kD and 64 kD proteins with difference in the N-terminal region [8]. The DNA replication origin and transcription enhancer motif are present at ~2kb upstream of the exon 1 in c-myc gene. The 7 nucleotide base pair motif sequence in this region is TCTCTTA and binds c-myc protein/c-myc protein complex. The motif is essential for the DNA replication and also transcriptional activation mediated by c-myc (H-P; HindIII-PstI site) region in Raji cells [9]. The translocation of c-myc genes often silences the untranslocated c-myc gene and activates translocated c-myc genes. However, some of the lymphomas such as Raji Burkitt’s lymphoma cells have both normal and translocated c-myc genes in transcriptionally active state but translocated c-myc genes are much more active. The translocation to chromosome 14 which is often found in Burkitt’s Lymphoma, and activates the c-myc gene transcription. The translocations are to CH region or JH region on chromosome 14. Minority translocations are occurring by t(8;2) and t(8;22) translocations. The common c-myc breakpoint is around exon 1. The tumors with breakpoint 5’ to exon 1 express normal c-myc mRNA of 2.25 kb and 2.4 kb with different 5’ ends from two different TSS (transcription start sites) [7]. However, tumors with breakpoint within exon1 or intron 1 express altered c-myc mRNA of 2.1–2.7 kb initiated from intron 1. The translation initiation site is located at nucleotide 16 of exon 2 and both types of translocations produce same c-myc proteins. The exon 2 encodes 252 amino acids and exon 3 encodes 187 amino acids. The intron1 is 1,616 bp long in human [10].The c-myc gene translocations in Burkitt’s lymphoma often lead to shortening or loss of the exon1 and multiple somatic mutations in exon 1. One noted example is BL22 cell line where the breakpoint is at up stream of 5’ end of c-myc gene, but there are mutations near the 3’ end of exon 1 close to donor site of splicing [7]. In the COLO 320 cell line, there are both normal and exon 1 truncated c-myc genes exist and both are expressed. It was found that the exon 1 truncated c-myc mRNA has much longer half-life than the normal c-myc mRNA which has half-life ~30 min [11]. Therapeutically, to inhibit the c-myc gene expression, the c-myc gene DNA, RNA transcrips and effector c-myc protein can be targeted. The c-myc protein is labeled as undruggable protein due to its structural instability and sought for the interruption of protein-protein (Myc-Max etc) interactions for the inhibition of c-myc activity. At the level of DNA, targeting to promoter 1 (P1) [12] and promoter 2 (P2) [13] for the transcriptional inhibition were made by TFO (Triplex Forming Oligodeoxynucleotides) or in combination of TFO with daunomycin for the triplex stabilization in HeLa cells. Antisense oligonucleotides therapy for the c-myc gene activity is focused on the protein expression targeting to mature c-myc mRNA.
These are all for the normal c-myc gene over expression disregarding mutation sites or changes in RNA sequences and structures. It was of interest to see how the RNA transcript of c-myc gene may change by mutations and translocations which can be targeted. The c-myc gene sequence is retrieved from GenBank: X00364.2 and analyzed potential secondary structure formations by RNA Fold program in Albany New York. The changes in BL22 Burkitt’s lymphoma due to mutations in exon 1 splice region was also analyzed. Overall the secondary structure in the mutated region changes to the less stable structure which may favor the spliceosome formation. However, utilizing two other RNA fold program such as Rochester program and Q-fold program in addition to New York program, the local sequence of 40 to 100 nucleotides at the exon 1 and intron 1 region was examined. The stable hair pin structure at the site with unknown function was noticed which may be targeted by antisense or antisense in combination with small molecular antibiotics.
Materials and Methods
The c-Myc gene sequence is retrieved from the GenBank:X00364.2. The sequence from 2327 to 7740 is utilized for the analyses. The exon 1 is from 2327–2881, intron 1 is from 2882–4505, exon 2 is from 4506–5277, intron 2 is from 5278–6653 and exon 3 is from 6654–7560 [one poly(A) site] or 6654–7740 [two poly(A) sites]. The 5’ UTR is from 2327–2881 + 4506–4520 (555 nt of exon 1 plus 15 nucleotides of exon 2), coding region is from 4521–5277 + 6654–7213 (757 nt of exon 2 plus 560 nt of exon 3) and 3’ UTR is from 7214–7560 [one poly(A) site included] or 7214–7740 [two poly(A) sites included] (Figure 1). The exon 1 mutation sequences of BL22 Burkitt’s Lymphoma is from the reference [7] (Figure 2). The UNA Fold Web Server is utilized for the RNA folding.
Figure 2. Changes in Sequence by Mutations in Translocated BL22 c-myc Gene from germ line c-myc gene at last 50 nucleotides of Exon 1

Results
Differences in Base Pairing Potentials in 5’ UTR, Coding and 3’UTR Regions
The base pairing potential of the sequence in the 5’ UTR is highest among coding region and 3’ UTR, and the 3’ UTR has the least base pair potentials (Figure 3 & Table I) in c-Myc gene transcript. These differences are also found in FMR1 gene transcript by different folding methods [14].
Figure 3. Modular RNA Secondary Structures of c-Myc mRNA
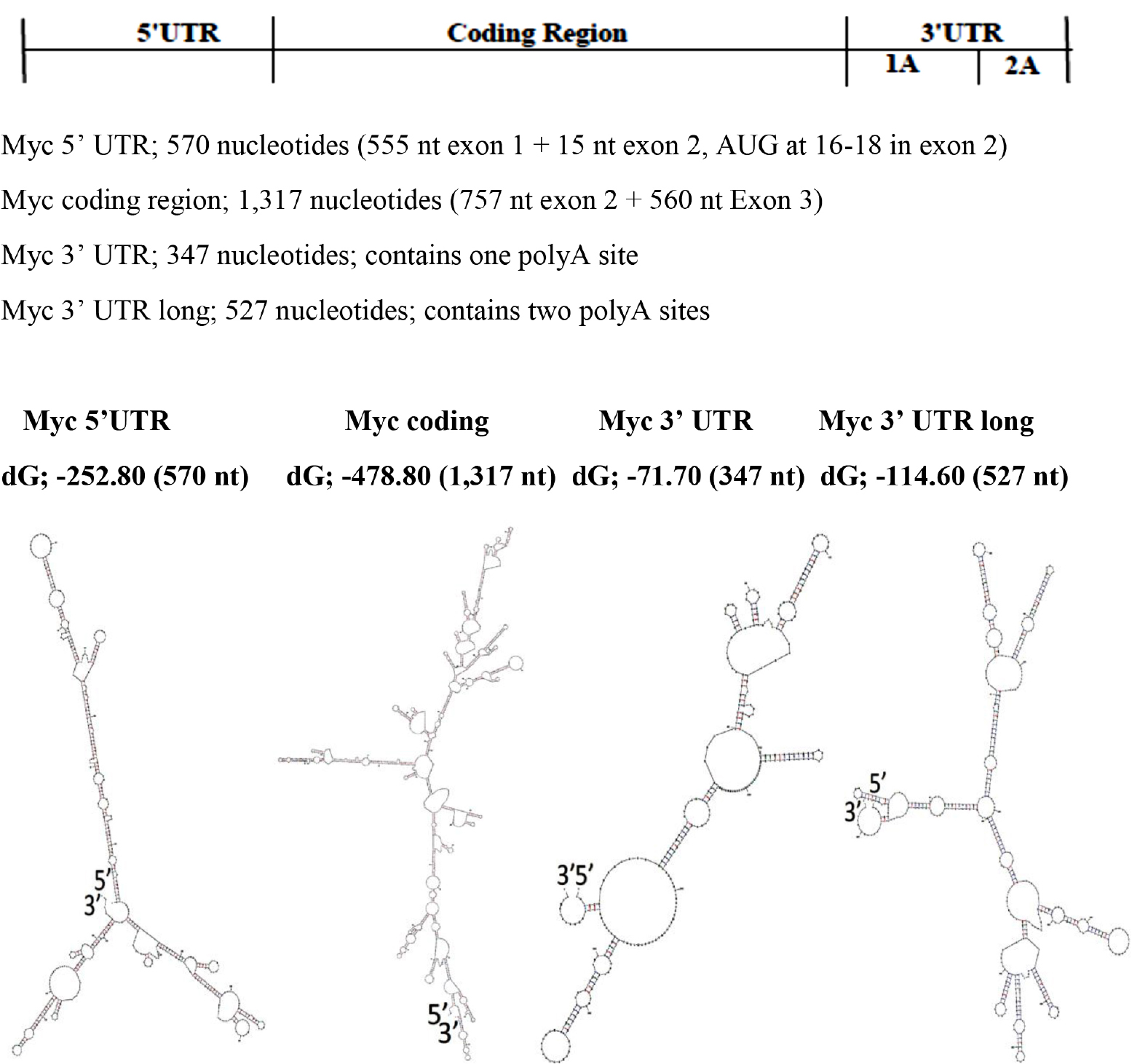
The RNA foldings are made using “The UNAFold Web Server” at the RNA Institute, Albany New York. The lowest dG Structures are Illustrated.
Table I. The free energy (dG) per Np (For the comparison, dG values are divided by number of nucleotides in the structure)
|
Region |
dG |
Length (Nt) |
dG/Np |
|
5’ UTR |
-252.80 |
570 |
-0.4435 |
|
Coding Region |
-478.80 |
1,317 |
-0.3635 |
|
3’ UTR 1pA site |
-71.70 |
347 |
-0.2066 |
Base Pairing Potentials in Exons and Introns
With regards to differences in base pairing potential between exons and introns, it was found that there are no significant differences but the base pairing potentials decrease towards 3’ end of the transcript.
(Figure 4 & Table II).
Figure 4. Modular Secondary Structures of pre-mRNA of c-myc gene Transcript
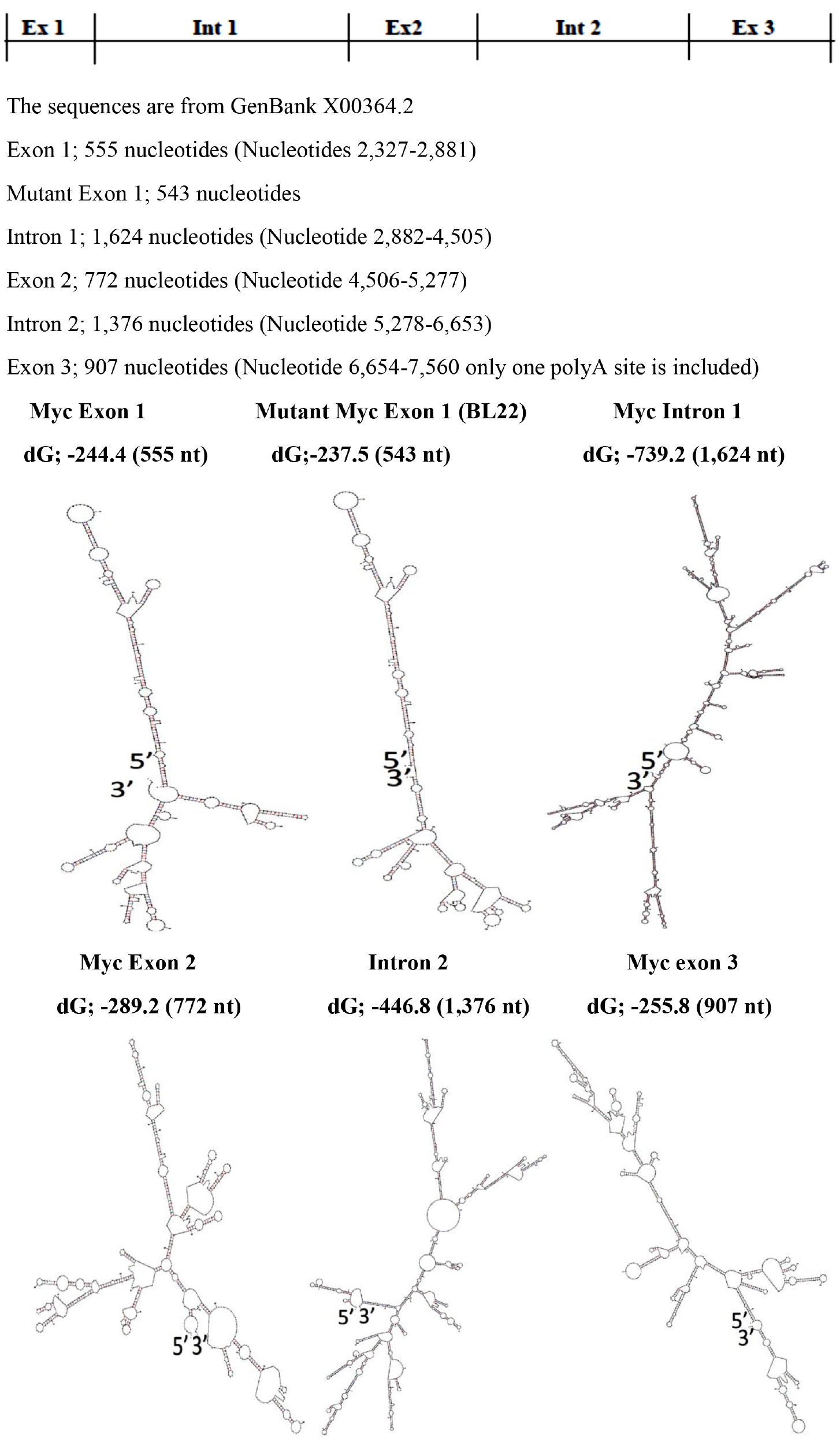
The Modular Secondary Structures are formed by using The UNAFold Web Server at the RNA Institute, Albany New York. The lowest dG Structures are Illustrated.
Table II. Base Pairing Potentials of Exons and Introns
|
Region |
dG |
Length (Nt) |
dG/Np |
|
Exon 1 |
-244.40 |
555 |
-0.4404 |
|
Intron1 |
-739.20 |
1,624 |
-0.4552 |
|
Exon 2 |
-289.20 |
772 |
-0.3746 |
|
Intron 2 |
-446.80 |
1,376 |
-0.3247 |
|
Exon 3 |
-255.80 |
907 |
-0.2820 |
Changes in Base Pairing by Mutations
The mutation sequences available to be found is mutations in the exon 1 of BL22 Burkitt’s lymphoma. Although the c-myc gene translocation break point is ~1000 bp upstream of the mutation area, the mutations are clustered at the 3’ end of exon 1 where the pause /block motifs are present which attenuates the pause/block function making the mutation more favorable for the read-through at the junction (Figure 5). The corresponding segment of untranslocated allele in BL22 was identical to prototypic wild-type sequence [7]. The mutation is less than 10% (37 nt out of 550 nt) and over all folding pattern is not much different from control sequences (Figure 4). Interesting folding pattern reveals the long double stranded helix by base pairing potential in exon 1 in both control and mutated exon 1 in comparison with other parts of the sequence. This may suggest that the region in exon 1 can be targeted by RNase III in favor of other parts which may be in concordance of rapid turnover of full length c-myc mRNA in comparison with truncated c-myc mRNA. The study of local region of mutated exon one reveals the different folding pattern which can be visualized that made more unstable structures at the splice site of mutated c-myc gene transcript in favor of easier spliceosome formation (Figure 5). The mutation also altered the U1 snRNA 5’ binding region (Figure 6) which is less favorable than the control in spliceosome formation. The mutation created long C-track close to splice site which may be targeted for the enhancement of the elongation block and inhibition of splicing. (Figure 2)
Figure 5. Changes in Secondary Structures at the Splice site by Somatic Mutations
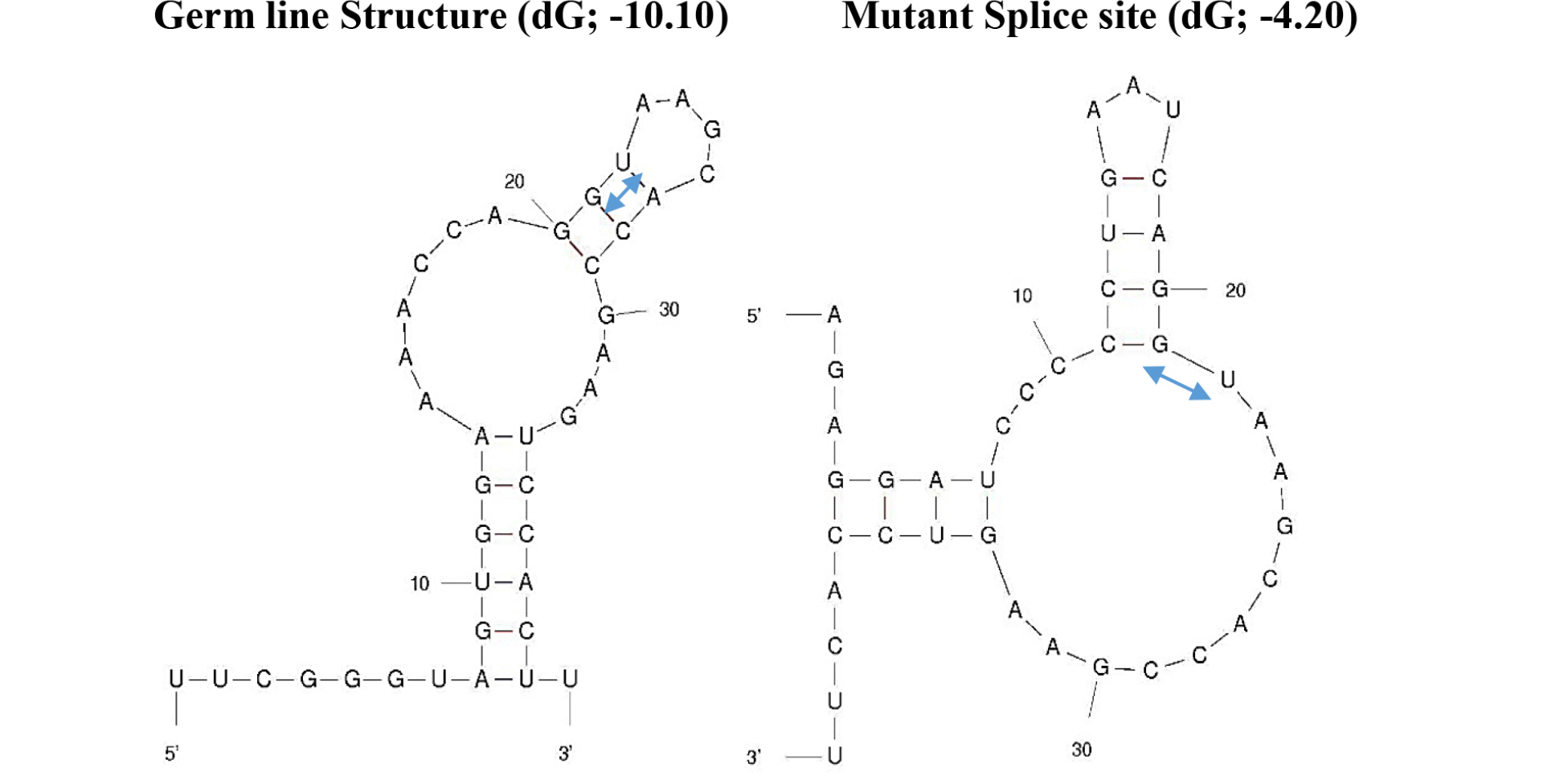
The secondary structures are made by UNAFold Web Server at The RNA Institue, Albany New York using sequences 20 nucleotides from 3’ end of exon 1 and 20 nucleotides from 5’ end of intron 1. The splicing motif –GU-s are marked by 
Figure 6. The Changes in Complementary Sequence to U1 snRNA 5’ Fragment
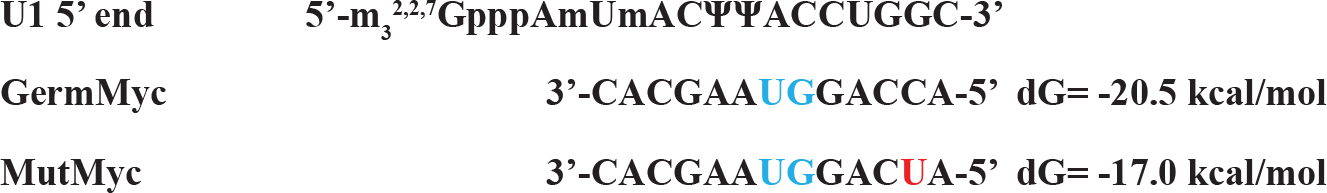
Discussion
The c-Myc protein is a site specific DNA (12 nt palindrome GACCACGTGGTC) binding transcription factor [15] and elevated in >15% of all cancer cells by gene amplification (double minutes), translocation, somatic mutations, activation of upstream of c-Myc gene and retroviral insertion. The c-myc affects all levels of gene expression at the transcription, splicing and translation. The myc protein is a transcription factor which in combination with other proteins enhances transcriptional activity of genes participating on cell division, growth and cell survival. The c-myc protein contains multiple transcription activation domains (TADs) in the amino terminal domain and recruits transcription cofactors and chromatin regulators. The c-Myc protein has been found to bind TRRAP (transformation/transcription domain-associated protein) which is scaffold protein and also binds E1A/E2F. The antisense to TRRAP abrogates cell transformation by c-Myc protein. The TRRAP is recruited by c-Myc to specific chromosomal site [16]. The TRRAP has revealed that it recruits histone acetyltransferase hGCN5 to c-Myc bound site for gene activation [17]. The c-Myc at the transcription, the myc protein function at the RNA polymerse II release stage at the promoter proximal pause site. The c-myc binds to E-box in promoters of many active genes and stimulates recruitment of elongation factor p-TEFb, [18] [19] [20]. Using the CAD gene, (Carbamoyl-phosphate synthetase 2, Aspartate Transcarbamylase and Dihydroorotase, trifunctional enzyme coding gene) and immune precipitation method, it was found that the CAD gene has RNA polymerase II bound form in quiescent NIH 3T3 and in differentiated U937 cells without promoter activity. The serum stimulation of the cell growth for the activation of the genes leads to phosphorylation of pol II CTD and its c-myc association which is detectable by antibody to hyperphosphorylted form of pol II CTD [19]. The c-myc also has effects on the splicing [21] [22] and translation [2] [23]. The c-Myc protein is a transcription factor which increases splicing factors such as SRSF1, Sam68 and introduces alternative splicing on cell cycle control proteins in favor of cell proliferation [21] [22]. In addition, splicing factor BUD3 is required for the c-myc driven cell proliferation. The knock down of BUD3 in c-myc hyperactive state reveals the increased intronic retention and cell apoptosis [24]. The inhibition of c-myc expression/function can be made by targeting transcription, splicing and translation. At the level of transcription, the inhibition of upstream regulatory element, inhibition of promoters have been demonstrated. The translational inhibition by antisense oligonucleotides at the AUG initiation region revealed significant inhibitory effects. However these agents are targeting normal gene over expression without specificity to cancer specific sequences which may arise by mutations or translocations.
In addition, the c-myc gene has regulatory elements at the end of exon 1 and intron 1 region where the ratio of exon1/exon2 changes in different genomic condition leading to differentiation or transformation of the cells. In many cases of Burkitt’s lymphoma, the mutation at the end of exon 1 increases the read through pause/block site and decreases the exon1/exon2 ratio, leading to in favor of cell proliferation. The mutations in BL22 lymphoma is consistent with this finding, where local duplication, T deletions and T/G to C mutations eliminate pause site and increases the read through of the pause site. At the transcript level, the changes in secondary structure make it more favorable for the spliceosome formation (Figure 5). This in turn can make accessibility of target site by antisense more favorable. Screening the local region (exon1/intron1), the hairpin structure was found which can be formed in both normal and mutated but in normal flanking sequence make the hairpin structure formation unfavorable (Figure 7). It may be feasible to target this mutated –CCCCC- sequence in combination of hairpin targeting small molecule for more effective therapeutic strategy. There are extensive study on inhibition of promoter function as well as inhibition of translation by antisense oligonucleotides. Examples are illustrated as below.
Figure 7. Alternative hairpin structures at the splice site between the exon 1 and intron 1
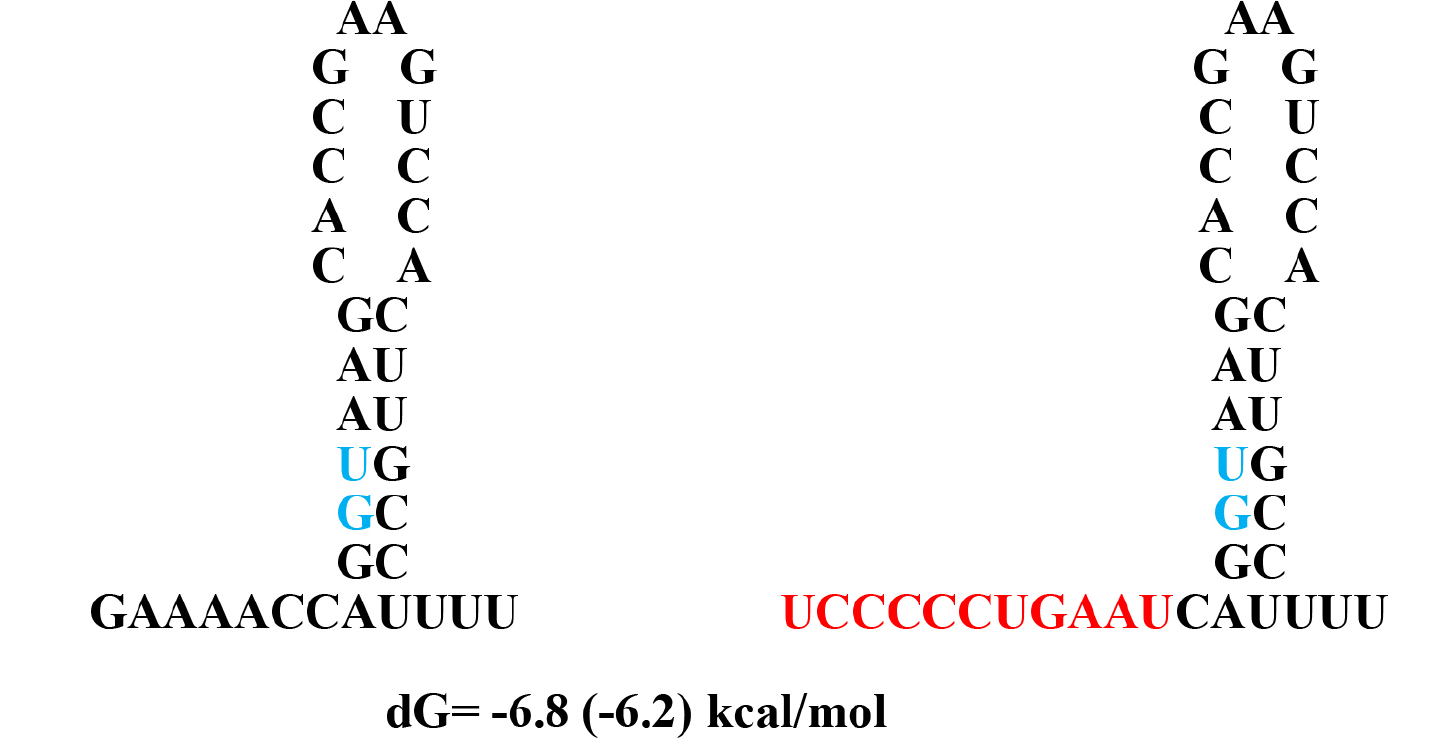
dG values are calculated with values in the table in the book by von Heijine, G. The number in parenthesis is from UNAFold Web Server which also makes same structure as Q-fold program.
A. The TFO (Triplex Forming Oligonucleotide) to P1 (Promoter 1)
The Target site -153 to-117 from +1 of P1
5’ –TCTCCTCCCCACCTTCCCCACCCTCCCCACCCTCCCC- 3’
3’ –AGAGGAGGGGTGGAAGGGGTGGGAGGGGTGGGAGGGG- 5’
Target sequence is colored in red.
PU1 TFO for the Target
5’ –TGGGGAGGGTGGGGAGGGTGGGGAAGG- 3’
Underlines are mismatch for the preferred antiparallel orientation for triplex formation.
Scrambled control ODN
5’ –CCTTCCCCACCCTCCCCACCCTCCCCA- 3’
B. The TFO to P2 site (triple helix forming oligonucleotides at P2)
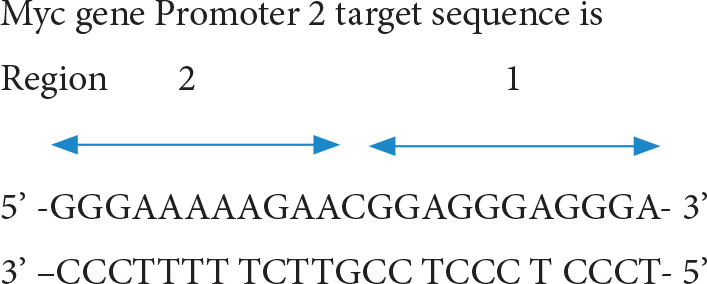
The short TFO against region 1 anti-parallel oligo; 5’ –TGGGTGGGTGG- 3’ and the TFO against region 2 parallel oligo 5’ –CCCTTTTTCTT- 3’ have been found to form stable triple helix and inhibits transcription factor binding. The longer triple forming oligo for full length also was effective in triplex forming and inhibition of protein binding [25]. Moreover, conjugation of daunomycin to TFO (dauno-GT11; dauno-TGGGTGGGTGG-3’) increased stability of its triple helix by intercalating target site DNA. The dauno-GT11 inhibited transcription of myc gene in vitro in prostate and breast cancer cell lines [26]. The combined TFO covering entire P2 region (3’ –TGGGTGGGTGGTTTGTTTTTGGG- 5’) in comparision with control scrambled TFOc (3’ –GGTGTGTGGTGTGGGTGGG- 5’) has been shown to inhibit COLO 320DM human colon cancer xenografts (containing amplified c-MYC)in mice [27].
C. Antisense to Translation Initiation Site;
Antisense phosphorodiamidate morpholino oligomer [28]
AVI-4126; 5’ –ACGTTGAGGGGCATCGTCGC- 3’
Target site 3’ –UGCAACUCCCCGUAGCAGCG- 5’
AVI-144 Scrambled; 5’ –ACTGTGAGGGCGATCGCTGC- 3’
antisense oligodeoxynucleotide [29]
as ODN 5’ –TAACGTTGAGGGGCAT- 3’
Target site 3’ –AUUGCAACUCCCCGUA– 5’
scrambled 5’ –TAAGCATACGGGGTGT- 3’
The initiation codon AUG is highlighted in blue and scrambled nucleotides are colored in red.
these antisense oligonucleotides have shown significant inhibition of cMyc synthesis in comparision with scrambled oliginucleotides
D. Potential target for the druggability.
The mutation sequence –CCCCC- in conjunction with stable stem-loop structure (Fig. 7) may serve as potential target for the lymphoma treatment.
It is an approach to find druggable site using computer screening the primary and secondary structure of RNA transcript at the mutation sites
References
- Marcu KB, Bossone SA, Patel AJ (1992) myc FUNCTION AND REGULATION. Annu Rev Biochem 61: 809–860.
- Choi H, Jackson NL, Shaw DR, Emanuel PD, Liu L, et al. (2008) mrtl-a translation/localization regulatory protein encoded within the human c-myc locus and distributed throughout the endoplasmic and nucleoplasmic reticular network. J Cell Biochem 105: 1092–1108.
- Krumm A, Meulia T, Brunvand M, Groudine M (1992) The block to transcriptional elongation within the human c-myc gene is determined in the promoter-proximal region. Genes Dev 6: 2201–2213.
- Eick D, Bornkamm GW (1986) Transcriptional arrest within the first exon is a fast control mechanism in c-myc gene expression. Nucl Acids Res 14: 8331–8345.
- Chung J, Sussman DJ, Zeller R, Leder P (1987) The c-myc gene encodes superimposed RNA polymerase II and III promoters. Cell 51: 1001–1008.
- Bentley DL, Groudine M (1986) A block to elongation is largely responsible for decreased transcription of c-myc in differentiated HL60 cells. Nature 321: 702–706.
- Taub R, Moulding C, Battey J, Murphy W, Vaaicek T et al. (1984) Activation and Somatic Mutation of the Translocated c-myc Gene in Burkitt Lymphoma Cells. Cell 36: 339–348.
- Hann SR, King MW, Bentley DL, Anderson CW, Eisenman RN (1988) A Non-AUG Translational Initiation in c-myc Exon1 Generates an N-terminally Distinct Protein Whose Synthesis Is Disrupted in Burkitt’s Lymphomas. Cell 52: 185–195.
- Ariga H, Imamura Y, Iguchi Ariga SMM (1989) DNA replication origin and transcriptional enhancer in c-myc gene share the c-myc protein binding sequences. EMBO J 8: 4273–4279.
- Bernard O, Cory S, Gerondakis S, Webb E, Adams JM (1983) Sequence of the murine and human cellular myc oncogenes and two modes of myc transcription resulting from chromosome translocation in B lymphoid tumours. EMBO J 2: 2375–2383.
- Rabbitts PH, Forster A, Stinson MA, Rabbitts TH (1985) Truncation of exon 1 from the c-myc gene results in prolonged c-myc mRNA stability. EMBO J 4: 3727–3733.
- Postel EH, Flint SJ, Kessler DJ, Hogan ME (1991) Evidence that a triplex-forming oligodeoxyribonucleotide binds to the c-myc promoter in HeLa cells, thereby reducing c-myc mRNA levels. Proc Natl Acad Sci USA 88: 8227–8231.
- Kim HG, Miller DM (1995) Inhibition of in vitro Transcription by a Triplex-Forming Oligonucleotide Targeted to Human c-myc P2 promoter. Biochemistry 34: 8165–8171.
- Ro-Choi TS, Choi YC (2007) A Modeling Study of Co-transcriptional Metabolism of hnRNP Using FMR1 Gene. Mol Cells 23: 228–238.
- Halazonetis TD, Kandil AN (1991) Determination of the c-MYC DNA-binding site. Proc Natl Acad Sci USA 88: 6162–6166.
- Mc Mahon SB, Buskirk HAV, Dugan KA, Copeland TD, Cole MD (1998) The Novel ATM-Related Protein TRRAP Is an Essential Cofactor for the c-Myc and E2F Oncoproteins. Cell 94: 363–374.
- Mc Mahon SB, Wood MA, Cole MD (2000) The Essential TRRAP Recruits the Histone Acetyltransferase hGGCN5 to c-Myc. Mol Cell Biol 20: 556–562.
- Rahl PB, Young RA (2014) MYC and Transcription Elongation. Cold Spring Harb Perspect Med 4 .
- Eberhardy SR, Farnham PJ (2001) c-Myc Mediates Activation of the cad Promoter via a Post-RNA Polymerase II Recruitment Mechanism. J Biol Chem 276 : 48562–48571.
- Eberhardy SR, Farnham PJ (2002) Myc Recruits P-TEFb to Mediate the Final Step in the Transcriptional Activation of the cad Promoter. J Biol Chem 277: 40156–40162.
- Das S, Anczuków O, Akerman M Krainer AR (2012) Oncogenic Splicing Factor SRSF1 Is a Critical Transcriptional Target of MYC. Cell Reports 1: 110–117.
- Caggiano C, Pieraccioli M, Panzeri V, Sette C, Bielli P (2019) c-MYC empowers transcription and productive splicing of the oncogenic splicing factor Sam68 in cancer. Nucl Acids Res 47: 6160–6171.
- Cargnello M, Topisirovic I (2019) c-Myc steers translation in lymphoma. J Experimantal Medicine 216: 1471–1473.
- Hsu TY, Simon LM, Neill NJ, Marcotte R, Sayad A et al. (2015) The spliceosome is a therapeutic vulnerability in MYC-driven cancer. Nature 525: 384–388.
- McGuffie EM, Catapano CV (2002) Design of a novel triple helix-forming oligodeoxynucleotide directed to the major promoter of the c-myc gene. Nucl Acids Res 30: 2701–2709.
- Carbone G, McGuffie E, Napoli S, Flanagan CE, Dembech C, et al. (2004) DNA binding and antigene activity of a daunomycin-conjugated triplex-forming oligonucleotide targeting the P2 promoter of the human c-myc gene. Nucl Acids Res 32: 2396–2410.
- Boulware SB, Christensen LA, Thames H, Coghlan L, Vasquez KM et al. (2014) Triplex-forming oligonucleotides targeting c-MYC potentiate the anti-tumor activity of gemcitabine in a mouse model of human cancer. Mol Carcinog 53: 744–752.
- Iversen P, Arora V, Acker AJ, Mason DH, Devi GR (2003) Efficacy of Antisense Morhpolino Oligomer Targeted to c-myc in Prostate Cancer Xenograft Murine Model and a Phase I Safety in Humans. Clin Cancer Res 9: 2510–2519.
- Pastorino F, Brignole C, Marimpietri D, Pagnan G, Morando A et al. (2003) Targeted Liposomal c-myc Antisense Oligodeoxynucleotides Induce Apoptosis and Inhibit Tumor Growth and Metastasis in Human Melanoma Models. Clin Cancer Res 9: 4595–4605.
- von Heijine G (1987) Nucleotide sequences: what you can do with your sequence once you have it. Sequence analysis in molecular biology (1stedn), Academic Press 19–80.