Abstract
The major obstacle of dermal and transdermal therapeutics is the low penetration rate of xenobiotics through the skin due to the diffusional barrier of its upper layer, the stratum corneum. A wide array of techniques has been proposed thus far, however, recent developments in the application of nanosystems for topical drug administration have gained much interest and optimism. In this review, we provide a comprehensive overview of the main types of nanocarriers that have been studied and developed up-to-date. We conclude that the nanosystems may become a useful dosage form for a variety of dermally active principals by modulating drug transfer and serving as nontoxic penetration enhancers.
Keywords
Microemulsion, Nanoparticles, Skin Permeation, Transdermal Drug Delivery, Nanotechnology.
Introduction
An optimal pharmaceutical dosage form aims at delivering an active compound to a target organ at therapeutic concentrations, while avoiding or reducing side effects and simplifying the dosing regimen of the patients. Percutaneous drug delivery is one of the promising pharmaceutical approaches since it can provide a continuous delivery for hours or days creating constant drug levels. The transdermal delivery can be advantageous in particular for drugs with a high renal or hepatic clearance, i.e., those that undergo oral first-pass metabolism and drugs with short half-lives that would otherwise need a frequent dosing regimen. The quick and short-acting BCS class II drugs with a very short half-life like the antidiabetic repaglinide, are ideal candidates for transdermal drug delivery [1]. In addition to the potential improvement in the medication regimen, the transdermal route can cause a considerable impact in medical practice, clinically and economically, by improving the treatment compliance and reducing the need for medical services. In an observational study of a population with Alzheimer’s disease [2], a high proportion of patients have switched from oral to transdermal rivastigmine, which has eventually resulted in increased patient satisfaction and stress relief for caregivers through the use of the patch under daily practice conditions. The ultimate goal, though not easy to fulfill, for all new forms of transdermal formulations is to achieve a quantitative drug permeation through the skin, taking into account that the skin is an excellent barrier and basically difficult to penetrate. This constraint is evidenced by the fact that after more than 30 years since launching the first transdermal drug product, there have been only 30 transdermal products on the US market for just 20 drug molecules [3,4]. Nevertheless, due to the advantages of transdermal drug delivery and the potential benefit for millions of patients every year, tremendous research efforts have been made to challenge the skin penetration problem. Many techniques have been developed to enhancing transdermal drug permeation using physical or chemical methods. The latter include prodrugs, salt formation, ion pairing, chemical enhancers, as well as nano-formulation approaches such as microemulsions, nanoemulsions, solid lipid nanoparticles, polymeric nanoparticles, nanostructured lipid, liposomes and others [5–10]. Microemulsions (MEs), and nanoparticles which includes lipid nanoparticles (SLNs and NLCs) and solid polymeric nanoparticles (PNPs), three nanotechnology-based dosage forms (see Table 1 and Fugure 2), have been drawn much attention during the last decade. Owing to their physicochemical properties, these systems are proposed as vectors to deliver active compounds through the stratum corneum, epidermis, and dermis, in order to obtain dermal, regional, and systemic effects. It should be noted that dermal and transdermal drug delivery systems are both being designed to overcome the main barrier of the skin, the stratum corneum, and the terminological difference between them relates to the extent of the molecular flux through the skin. Unfortunately, due to the selective nature of the stratum corneum, only a small group of drugs loaded in these novel formulations can be delivered at a therapeutically relevant dosage [11]. In this review, we summarize the immense work done so far aiming to challenge the skin barrier by the three types of nano-sized drug delivery systems.
Table 1. MEs, PNPs and SLNs for drug delivery to and through the skin.
Topical Pharmaceutical Forms and Challenges in Transporting Drugs via the Transdermal Route
Since the US Food and Drug Administration have approved the first scopolamine transdermal patch for motion sickness in 1979, the pharmaceutical industry has centered their efforts on transdermal formulation research for other drugs. Between 1970 to 1980, the first generation of the transdermal system included patches that designed as a drug reservoir with a semi-permeable membrane placed over the skin that controls drug release. Later, in the next decade (1980–1990), patches made of polymeric matrices entrapping the drug within pores and channels were introduced into the market. Today, almost 20 drugs have been successfully incorporated into transdermal drug delivery systems to reach systemic circulation or to treat local disorders. Commercial transdermal systems contain drugs such as clonidine, fentanyl, estradiol, nicotine, rotigotine, estradiol, oxybutynin, testosterone, nitroglycerin, and others [3]. The transdermal dosage forms have become attractive for patients due to their simplicity, immediate application, and prompt termination whenever desired [12–15].
Advantages of Transdermal Drug Delivery
Transdermal drug delivery has a significant advantage of bypassing the hepatic first-pass metabolism, thereby improving drug bioavailability. In addition, the transdermal route can avoid possible gastrointestinal degradation and instability of some orally- administered drugs and/or unwanted mucosal irritation caused by drugs like non-steroidal anti- inflammatory drugs (NSAIDs). The transdermal benefits were exemplified in a research performed with flurbiprofen, one of the most potent members of the phenylalkanoic acid series of NSAIDs [16]. After its oral administration, the most frequently reported side effects of flurbiprofen are abdominal discomfort, diarrhea, constipation, emesis and abdominal distention [17]. Furthermore, it has a short half-life of 3.9 h, which requires frequent dosing that obviously increases these symptoms. These drawbacks make flurbiprofen a perfect candidate for the transdermal route, especially for the prolonged therapy of rheumatoid arthritis and its related disorders. Charoo et al. [18] have shown that the bioavailability of flurbiprofen in albino rats increased up to 5.56 times with transdermal patch formulation compared to its oral administration. The results were confirmed by pharmacodynamic studies in a rat edema inflammation model [18]. Another attractive advantage of the transdermal drug delivery systems is the reduction of toxic side effects by keeping a steady state plasma drug level with less peak-to-trough variations. The necessity of the transdermal route is also exemplified by the scopolamine patch that was designed to prevent motion sickness for an extended duration of time. Its dermal application accompanies with none or minimal adverse effects, which are usually associated with the oral or parenteral bolus therapy of antimuscarinic drugs (mainly dry mouth, drowsiness and some more reactions including hallucinations) [19, 20]. Finally, the improved patient compliance due to non-invasive, easy drug administration, and the natural capability of controlled and sustained drug release make this dosage form attractive. It may also effectively serve the elderly population, especially people suffering from dysphagia and neurological disorders that lead to non- compliance [21]. Similarly, it may serve infants and children who also require compliant medications [22]. As it has already been recognized that drug compliance is quite substantial, the transdermal drug delivery can resolve the compliance problems, reducing the need for injections and oral administration of medications to vulnerable pediatric and geriatric patients [23, 24]. In developing countries, children, unfortunately, die from easily treatable diseases (e.g., malaria) due to poor compliance, e.g., difficulties to properly swallow medications or inconsistent multiple dosing. Poor patient compliance can also result in antibiotic resistance, which is one of the biggest threats to global health.
Limitations of Transdermal Drug Delivery
Despite the considerable advantages some major drawbacks limit the transdermal patch from being a generally-used drug delivery system. A substantial limitation of the transdermal route is a poor permeation of many active molecules through the skin, wherein the stratum corneum plays the primary barrier. A relatively high skin penetration usually takes place with low molecular weight drug molecules (< 500 kDa), light lipophilicity, and high potency (daily dose < 10 mg) [25–27]. Due to these limitations, it is therefore difficult to frequently employ the transdermal route. Thus, it is a great challenge and almost an impossible mission to deliver large hydrophilic molecules (i.e., high molecular weight/poor lipophilicity) such as antibodies, peptides, proteins, and hormones. Besides, possible skin irritation and sensitization caused by the formulation and/or the adhesive band may be detrimental during the treatment. It should be taken into consideration that a drug that accumulates in the skin may be irritant if it stays for a relatively long time inside the skin layers. Therefore, it is essential to investigate the skin pharmacokinetics not only during the patch application but also after the application is over (patch removal). An excellent example of skin toxicity by accumulation is captopril, which its irritation activity could be diminished only by including penetration enhancer, antioxidant, anti-irritant and chelating agent in the transdermal formulation [28]. Inter- and intra-subject variability of skin permeability due to patient skin conditions may also be a problem. Fentanyl, a potent synthetic opioid that has been used in transdermal drug delivery systems, has caused an extensive interindividual variation in dermal penetration with maximal fluxes ranging between 21 to 105 ng/cm2/h in in vitro studies [29].
Finally, it is well known that the most common drug-metabolizing enzymes are expressed in the skin, which is the largest organ of the human body. Among them are cytochromes P450, flavin monooxygenases, glutathione-S-transferases, N-acetyltransferases, and sulfotransferases [30, 31]. Their activity in the skin may cause either drug biotransformation that leads to inactivation [32], or prodrug biotransformation that leads to pharmacological activation [33, 34]. Any pre-systemic metabolism that may occur in the skin should be taken into account when transdermal patch is designed and developed.
The Skin Barrier
Skin is the largest and accessible organ of the body, covering a surface area of approximately 2m2 depending on the individual weight and height. It is also one of the most multifunctional organs, forming a protective barrier against many different factors including ultraviolet radiation, pathogens, and xenobiotics. It allows, however, an exchange of gases and toxins with the external environment. Additionally, it prevents water loss but regulates body temperature in humans through sweat secretion. The thickness of the skin is a few millimeters, possessing two primary structures:
- The epidermis, an avascular layer, measures 50 to 100 µm in thickness. This layer can be further divided into four distinct strata according to the corresponding cell differentiation: stratum basale, stratum spinosum, stratum granulosum, and the outer stratum corneum (SC). The latter is the outermost layer, which is 10 to 20 µm thick and is generally accepted as the primary membranal barrier for topically applied xenobiotics. This barrier is constructed as a ‘brick and mortar’ structure, where the bricks represent dead corneocyte cells composed primarily of cross-linked keratin and the intercellular mortar is a mixture of lipids organized in bilayers. The extracellular medium consists principally of neutral lipids such as cholesterol, ceramides and fatty acids, including linoleic acid that is deemed to play a significant role in the barrier function [35]. The SC is commonly recognized as the most predominant barrier for drug delivery, therefore, it has become the target for variously designed nanocarriers. By transporting the SC, the drug could be released from its carrier and retained in the skin, known as topical drug, or could be dragged through the skin into the blood, an action known as transdermal drug. Whatever is the mechanism of drug release, the act of transporatation through the SC barrier by the nanocarrier is of great importance for transdermal researchers.
- The dermis, covered by the epidermis, possesses 1–2 mm thickness and contains a highly capillary area just below the dermal-epidermal junction. Drug molecules that reach the dermis are available for systemic drug absorption via the capillary area. The dermis represents another active layer of the skin that holds the hair muscles, blood supply, sebaceous glands, and nerve receptors.
Although the skin consists of various membranal layers and cell types, it is acceptable that the stratum corneum layer is the one that controls the entrance of xenobiotics, and despite its incredible thin thickness it provides an effective barrier for maintaining homeostasis [36]. The passage of a xenobiotic through the skin can follow three diffusive pathways (Figure 1): transcellular (intracellular), paracellular (intercellular), and appendageal (through eccrine sweat glands or hair follicles). The transepidermal pathway (intracellular and intercellular) requires crossing through the stratum corneum, i.e., corneocytes and lipid-dominant extracellular lamellar membrane structures. Polar drugs following the intracellular route can cross through corneocytes, while apolar drug molecules undergo intercellular transport through the continuous lipid matrix. The transappendageal route comprises a passage of molecules through the hair follicles or sweat glands; however, it is considered as a less significant route for most drugs [37].
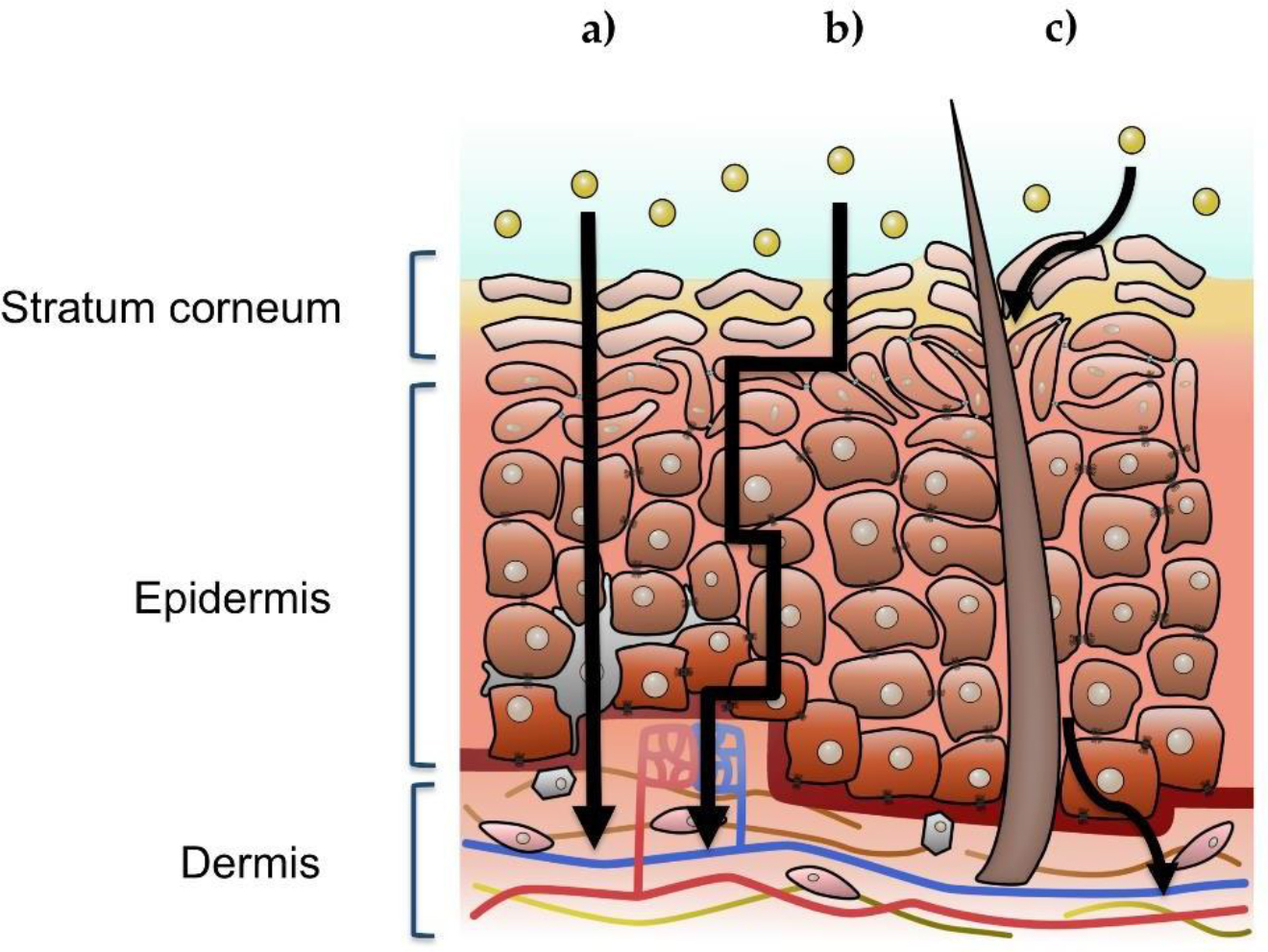
Figure 1. Illustration of a cross-section view of human skin, where the three possible xenobiotic penetration pathways are signaled: a) intracellular, b) intercellular, c) follicular or appendageal. The upper left inset indicates the stratum corneum, viable epidermis, and dermis.
Commonly, to obtain quantitative transdermal drug absorption to the dermis and blood circulation, a temporary disruption of the skin barrier function is required [38]. This process can proceed in three different steps. Initially, the molecule enters into the outer skin stratum, the SC (i.e., skin penetration). In the next step, it passes through one stratum to another, which is defined and known as permeation, and finally reaches the dermis and the vascular system (i.e., absorption). The physicochemical characteristics of the molecules determine the route of drug penetration through the stratum corneum [39]. It has been reported that hydrophilic molecules usually prefer the intracellular (transcellular) route, due to the low affinity of these molecules to lipids existing on the surface and between the corneocytes [40]. However, this pathway is considered to be most difficult due to the complexed lipid bilayers crossing. Moreover, if a drug is very hydrophilic, it would be unable to partition from the topical aqueous delivery system into the SC. In contrast, a highly lipophilic drug is unable to transfer into the epidermis and will retain in the SC. Like the intracellular route, the appendages are also considered as a less significant route, because they occupy a limited surface area (~0.1%) [41]. Altogether, the relative importance of the skin appendages depends on the intrinsic physical properties of the test molecules or delivery systems as well as the time of application [42]. Nevertheless, the appendages represent important reservoir structures and are considered as shunt penetration pathways [43]. In fact, the pilosebaceous units are key anatomic compartments for particle-based drug delivery systems. Nanoparticles in particular can be accumulated in the follicular openings and penetrate along the follicular duct [44, 45]. However, the hair follicle types and dimensions significantly vary among the different body regions, which make the delivery rate and extent via this route inconsistent in nature. It has been noted that while terminal hair follicles of the scalp and the dilated acne pores can conveniently be reached with particles in the submicron size range, penetration in hair follicles of the body vellum is limited to smaller particles sizes [46]. Despite its high tortuosity, the intercellular (paracellular) pathway of xenobiotics along the lamellar lipids between the corneocytes is widely considered as the main route for a few drugs but a significant barrier to permeation for most drugs. Therefore, it is commonly accepted that an optimal drug candidate for transdermal delivery should be lightly lipophilic (log octanol-water partition coefficient or logP = 1–3), possessing a low molecular weight (MW <500 Da), and a low melting point (m.p. <200 °C) [25, 47]. Basically, the intercellular and follicular route, at least in healthy skin, may be the most relevant for penetration of drug-loaded nanoparticles into the skin, but not for penetration of most drug molecules in their free and soluble form. Impenetrable large particles, rather than nanoparticles, deposited in furrows atop the stratum corneum may continuously release their payload, which may become effective only if the active molecules show suitable features for skin penetration such as described above.
Currently New Transdermal Drug Delivery Nano-Systems
There has been always of great importance for the pharmaceutical and cosmetic researchers to develop more “cost-effective/ delivery-efficient” vehicles. In particular, major efforts have been made for development of vehicles possessing an appropriate ability to carry hydrophilic and high molecular weight active molecules through the stratum corneum, molecules that are naturally impenetrable into the skin. These vehicles should be designed to bring about efficient penetration and maximization of drug bioavailability thus providing therapeutic drug concentrations. During the last decades, extensive research has been done to overcome the skin barrier and novel formulations and techniques have been developed to achieve this goal. They can be roughly classified into physical and chemical methods. The physical or the active methods include iontophoresis [48], electroporation [49], sonophoresis [50], fractional ablation [51], and micro-needles [52], and are used as a driving force to obtain drug skin permeation from a topical formulation. The iontophoretic method applies an electrical field to drive ionized drug molecules across the skin membrane, electroporation treats the skin with a high electrical voltage for a short period of time, and sonophoresis uses ultrasonic waves to improve drug diffusion. On another hand, there are chemical strategies that include chemical permeation enhancers that perturb the skin structure [53], prodrugs, and nanoformulations (or nanostructured systems) such as polymeric nanoparticles, solid lipid nanoparticles, and microemulsions (see Figure 2). Nanostructure systems represent an alternative to the traditional formulations due to their ability to facilitate drug delivery to structural features of the skin like hair follicles or interact with skin lipids to mediate transportation. Diverse parameters affect the penetration of nanostructures including size, shape as well as their deformability. The relevance of the size for nanoparticle permeation has not yet been conclusive, and that is because several other physicochemical properties of the nanoparticles may also affect, such as ζ potential and surface modification.
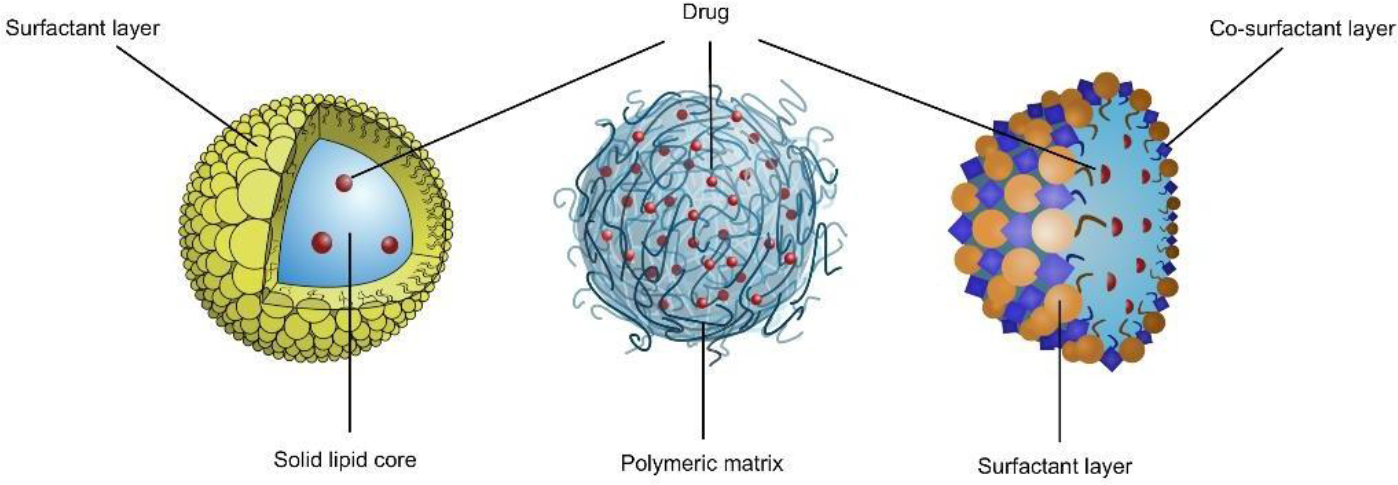
Figure 1.Figure 2. Schematic illustration of (from left to right): Solid lipid nanoparticles, Polymeric nanoparticles, and Microemulsion.
Microemulsion
Microemulsions (MEs) have gained an important role since it has widely demonstrated to significantly enhance transdermal permeation of drugs compared to the conventional dosage forms such as simple gels, ointments or creams (macroemulsions) [54]. A microemulsion is a mixture of two immiscible liquids, surfactant, and a cosurfactant, forming an optically isotropic structured liquid. Even though it macroscopically appears as a single-phase system, it is a multiphasic system on a nanoscale, being considered as a dispersion of nanodroplets. MEs are thermodynamically stable (interfacial tension is nearly zero) and are spontaneously formed while not requiring an input of energy. The ME formulation is beneficial for transdermal and dermal delivery of drugs, primarily since the diffusivity and the partitioning into the skin is significantly increased due to its high capacity, namely, a high quantity of drug that can be incorporated in the formulation. Furthermore, the microemulsion ingredients that create its unique composition may reduce the diffusional barrier of the stratum corneum and enhance skin permeation [55–58]. For instance, the hydration effect of the microemulsion on the stratum corneum has been reported to affect skin permeation [7,59]. In addition, ME formulations are capable of solubilizing and delivering both hydrophilic (in W/O MEs) and lipophilic drugs (in O/W MEs). Consequently, many studies have demonstrated an improvement in drug bioavailability and stability by using MEs. Gannu et al. [60] developed a microemulsion-based transdermal therapeutic system for lacidipine, which is a poorly water-soluble (logP=4.5) and poorly bioavailable drug. The results demonstrated a 3.5 times improvement in the bioavailability of lacidipine after transdermal administration of microemulsion gel compared to oral suspension. Other promising results have been reported by Said et al. [61] who studied the delivery of agomelatine by the transdermal route using microemulsions. An approximate value of 40-fold enhancement in drug permeation through rat skin was demonstrated when compared to the same drug in a hydrogel formulation.
Despite their benefits, MEs are not exempt from disadvantages. In order to produce stable nanodroplets with an ultra-low interfacial tension, high concentrations of surfactants (30–60%) are usually required. These excessive concentrations of surfactants may lead to skin irritation and toxicity. Additionally, due to the use of high concentrations of surfactants and co-surfactants, microemulsions can be affected by environmental parameters such as temperature, ionic strength, dilution, and pH. The effectiveness of MEs for transdermal drug delivery depends on several interconnected factors such as the microstructure, type, size, and shape of the nanodroplets. The transport efficacy through the skin layers also depends on how a drug or an active compound is incorporated in the MEs droplets, either dissolved in their core or attached at the interface. It has also been reported that MEs components can play an essential role as penetration enhancers, by disrupting the lipid structure of the stratum corneum or increasing the partition coefficient between the vehicle and the skin [62]. However, the penetration mechanism is additionally dependent on the stratum corneum properties and the physicochemical properties of the active compound [63]. Hathout et al. [64] have designed a study to evaluate how a microemulsion component induces changes in the stratum corneum. It has been shown that there was a proportional relationship between the perturbation degree of the stratum corneum and the concentration of the components that had topically applied on the skin [65]. They have shown that the use of unsaturated fatty acids, such as oleic acid, can decrease the conformational order of the stratum corneum lipids and induce some phase separation. Other studies have confirmed that the stratum corneum uptake of oleic acid, Tween 20 and Transcutol®, which are commonly used as MEs components, is increased after application of MEs compared to application of the pure chemicals [64]. Another interesting research evaluated the transdermal delivery of indomethacin in eugenol microemulsion by the rabbit ear model [66], showing a greater flux compared with that obtained from a saturated aqueous solution. This penetration enhancement can be explained by the the activity of eugenol and the synergistic effect obtained with PEG. Also, the microemulsions that contained a high eugenol concentration and Polyethylene Glycol (PEG) were able to increase the loading capacity of indomethacin as evidenced by a significant improvement in the solubility of the drug in these formulations compared to its water solubility (logP of indomethacin=4.46). In another study, ME was prepared with cineole as a chemical enhancer, a combination that promoted a higher skin permeation of zidovudine [67]. Zidovudine, a poorly bioavailable drug with a short half-life of 0.53h, significantly permeated through pig ear skin and snake skin from the cineole containing ME (by three-fold) compared with the control. It is well known that permeation enhancers may act on the skin through diverse mechanisms, such as by disruption of the lipid bilayer of the SC, or by interaction with skin proteins (e.g., denaturation, conformational modification). Specifically, cineole disorganizes the lipid bilayers and decreases SC property as a barrier.
Microemulsions can also act as skin permeation promoters by their nanostructural virtue. In a series of studies performed at Ben-Gurion University in Israel, the advantage of a microemulsion system has been established with no need of penetration enhancers or alcohols [8, 68–70]. The skin bioavailability of lidocaine was improved by a microemulsion system composed of glyceryl oleate and polyoxyl 40 fatty acids (as the surfactants), isopropyl palmitate as the oil, and tetraglycol as the co-surfactant [68]. The in vitro transdermal permeation of lidocaine was significantly increased from the microemulsion compared to lidocaine permeation from a macroemulsion, oil-free micellar system, and a surfactant mixture only (water-free). Similarly, the transdermal administration of diclofenac-containing microemulsion to rats resulted in 8-fold higher plasma levels of the drug than those obtained after application of Voltaren Emulgel® [8]. The diclofenac microemulsion contained the same ingredients as in lidocaine formulation [68], i.e., glyceryl oleate and polyoxyl 40 fatty acid, isopropyl palmitate tetraglycol. Caffeine permeation across fresh skin excised from rat, rabbit, and pig was highly enhanced when formulated in microemulsion composed of Labrasol®, glyceryl oleate, isopropyl palmitate, propylene carbonate and water [69]. The transdermal permeation rate of caffeine was higher via microemulsion than the rates measured after caffeine in Labrasol® solution, caffeine in an oil-free micellar system, and caffeine in a surfactant-oil mixture. These findings indicate once more that the mechanism of drug permeation through the skin is based on the microemulsion’s vesicular nature rather than on chemical enhancement driven by its surfactant excipients. In another study which was carried out by our group to evaluate transdermal curcumin permeation [70], it has also been established that microemulsion vehicle was advantageous over a micellar system and a surfactant-oil mixture, composed of the same proportions of ingredients.
Polymeric Nanoparticles
The use of polymeric materials for entrapping drugs and active substances in a solid envelope is an apparent approach selected to mask the intrinsic physicochemical properties of active compounds that are inapplicable for skin permeation. Polymeric nanoparticles, which are defined as nanosystems with particle sizes of tens and hundreds of nanometers in diameters, have frequently been given more descriptive names, such as nanocapsules or nanospheres, depending on their morphology. The presence of an inner core (aqueous or oily solution) in nanocapsules leads to a vesicular structure while its absence in nanospheres provides a matrix structure of the polymeric matter. The drug can be entrapped in the core (nanocapsules) or the channels and cavities of the matrix (nanospheres) as a dispersion (in a saturated solution), as a solution, or as a complex with the polymer. Due to their inherent complexity, nanoparticles made of polymers have shown to be an excellent carrier for controlled and sustained delivery of drugs [71, 72]. Their surface may also be modified in order to carry out an active or passive drug delivery [73, 74].
The pharmaceutical properties of polymeric nanoparticles (PNPs) (e.g., drug stability, release mechanism) depend essentially on the type of the polymer. For example, instability has been encountered in calcium alginate nanoparticles that led to fast and uncontrolled drug release profiles after oral administration [75]. Similar results have been shown in gliadin nanoparticles [76]. The proteinaceous nature of gliadin makes its nanoparticles highly sensitive to pH changes or ionic strength alterations, so they may dissolve or aggregate depending on the environmental conditions in the alimentary canal [76]. To improve PNPs’ stability, two approaches or strategies have been adopted. One of the strategies takes advantage of the functional groups present on the polymeric chains, which can be covalently cross-linked with a compatible crosslinker. In many cases, crosslinking is an essential step that affects the functionality of nanoparticles, such as biodegradability and/or drug release [77, 78]. The second strategy uses coating of PNP surface area (e.g., with polysaccharides, polyethylene glycols) to avoid agreggation and precipitation [79, 80]. PNPs are among the most studied nanocarriers for drug delivery due to their relatively high entrapment yield and their ability to effectively deliver therapeutic doses while minimizing side effects. Masella et al. [81] have developed an innovative polymeric patch for transdermal delivery of melatonin, which was incorporated into polycaprolactone (PCL) nanoparticles. Melatonin release from this patch was assessed by a Franz diffusion cell system, shown a controlled behavior of melatonin diffusion from the PCL nanoparticles. This drug has been indicated as a good candidate for transdermal drug delivery due to first-pass metabolism resulting in low oral bioavailability and a weak sleep-promoting effect. Another drug loaded in PNPs that has also been studied for transdermal delivery was pirfenidone, the first antifibrotic agent approved by the FDA to treat idiopathic pulmonary fibrosis. Pirfenidone has a short half-life in the (2.4 h), and clinical studies have shown that it undergoes hepatic first-pass metabolism. Moreover, following oral administration, it may cause side effects such as stomach pain, vomiting, diarrhea, burning or pain in esophagus and throat. Transdermal delivery has been attested as an effective pathway to overcome the undesired side effects, and to provide patient compliance. The transdermal drug delivery system was based on chitosan-sodium alginate nanogel, which presented a sustained release pattern during 24 h and a significant enhancement of skin penetration [82].
Lipid Nanoparticles (LN)
Solid lipid nanoparticles (SLNs) were introduced in 1990 as an alternative carrier system for liposomes, O/W microemulsions, and lipophilic polymeric nanoparticles. They are composed of a solid hydrophobic core and a monolayer of surfactant coating and are suspended in the aqueous environment. The solid core contains the active compound dissolved or dispersed in a solid high melting fat matrix. Due to their properties, SLNs have the potential to carry lipophilic or hydrophilic molecules, depending on the method of their preparation. Usually, SLNs are composed of approximately 0.1 – 30% (w/w) solid fat, have an average size of 50–1000 nm in diameter and a spherical morphology. Apart from decreasing SLNs’ particle size, which increases their stability, tensoactives are also being used at concentrations of about 0.5 to 5% to enhance stability. These include phospholipids, steroids, poloxamers, and polysorbates. The type of surfactants, lipid compounds, and their proportions can modulate the particle size and drug loading. SLNs’ lipid components include glyceryl esters, waxes, and fatty acids, which are required to be in a solid, state at ambient and body temperature (36.5–37.5°C). SLNs have several advantages: the manufacturing is cost-effective, easy to scale-up the production, they are biodegradable, relatively stable and nontoxic [83–851]. Other important characteristics include good protection offered for the entrapped drugs and sustained drug release from the lipidic matrix. For the purpose of transdermal application and to avoid a potential systematical toxicity, Guo et al. [86] prepared ivermectin-containing SLNs. The release study displayed a slow and sustained release patterns for the drug-SLNs. Nevertheless, variability was noted in the shape and particle size of the SLNs, as well as drug expulsion from the lipid matrix [87]. However, one of the most relevant limitations is the possible degradation of active components during the production process [88, 89]. Labile molecules such as peptides, proteins or nucleotides, may undergo degradation as a consequence of stress and strain caused by the homogenization process, or by the heat formation during melting. It should be aware, therefore, that an appropriate selection of the production method is indispensable. Gallarate et al. [90] prepared peptide-loaded SLNs through coacervation technique, a solvent-free method, in which leuprolide and insulin had been chosen as model polypeptides. The researchers have demonstrated that the coacervation technique, which included some mild heating steps, did not affect the chemical stability of these peptides.
SLNs appear to be an attractive colloidal carrier system for the delivery of drugs into the skin, mainly because of their soothing effects on the skin. They have a moisturizing effect on the skin through occlusion providing an incremental skin hydration [91]. SLN formulations were loaded with lornoxicam, an NSAID, and were tested for drug permeation through full-thickness rat skin [88]. As already mentioned, topical application of NSAIDs may represent drug administration that avoids some gastrointestinal side effects such as dyspepsia, ulceration, and bleeding, usually appearing after oral lornoxicam. It was shown that lornoxicam SLNs increased skin permeation rate compared with a lornoxicam gel control, implying that the spontaneous occlusivity and skin hydration increase the penetration into the skin [92]. Transdermal drug delivery using SLNs has also been studied for rivastigmine, a drug used for the treatment of mild to moderate Alzheimer’s and Parkinson’s diseases [93]. The results showed improvement of daily activities like cognition, behavior, and global function, thus, transdermal drug delivery have given an optimal advantage to such patients, by providing a controlled drug release that maintains steady plasma levels. That makes the treatment a user-friendly and convenient alternative to the traditional dosage forms. The researchers also reported that SLNs were prepared by the emulsification-diffusion method, and subsequently, the SLNs were incorporated into transdermal films. Then, pharmacokinetics studies were performed on rabbits, showing significant improvement of Cmax and bioavailability of the drug compared to a control patch [93].
SLNs can be modified by incorporation of liquid lipid into the solid structure. This new form of SLNs, named nanostructured lipid carriers (NLCs), encounters the limitations of SLNs such as limited drug loading capacity and lipid crystallization [95]. The introduction of liquid lipids causes a melting point depression in comparison with a pure solid lipid, and impairs the crystal structure of the lipid thus offering more space to drug inclusion. However, Teeranachaideekul et al [96] have shown that lipid NPs display a deeper penetration up to upper dermal layer when the content of liquid lipid is lower, indicating that liquid lipid limits skin penetration. This phenomenon has been explained by reduction in the occlusive effect (hydrophobic film formation) of the lipid nanoparticles with the increase on liquid content, which in turn caused more water evaporation and lesser skin hydration [96].
MEs, PNP and LN as Nanocarriers for Transdermal Drug Delivery of Various Therapeutic Agents
During the last decades, researchers have shown an increasing interest in nanocarriers for transdermal application. Table 1 summarizes the latest studies related to transdermal nanocarriers. The selection of the most appropriate carrier has been primarily dependent on the physicochemical characteristics of the drug that has to be loaded (e.g., pKa, hydrophilicity, lipophilicity), and factors related to the carrier and its manufacturing process, such as the ability to entrap the drug and to keep its stability. The nanocarriers could be prepared from a diversity of materials such as lipids, proteins, polysaccharides, natural or synthetic polymers. The lipophilic/hydrophilic nature of the drug mainly dictates the development of most compatible transdermal dosage forms. It is commonly accepted that drugs with a relatively high logP are more appropriate to be delivered through the skin, while drugs with low logP are more difficult to penetrate and need lipophilic carriers. Therefore, it is essential for a nanocarrier not only to be able to adsorb and entrap a low logP drug but also to penetrate and deliver it through the lipid bilayers of the stratum corneum [97].
Microemulsions have gained much interest due to their thermodynamic stability and ease of preparation. In vitro [113] and in vivo [114] studies have demonstrated their potential in enhancing drug delivery through dermal and transdermal routes of administration. The main advantages of MEs for transdermal delivery of drugs include (a) the high capacity and ability to solubilize quantitative amounts of hydrophilic drugs, and (b) the permeation enhancing effect due to individual components in the microemulsions or due to the vesicular nature of the delivery system. El Maghraby et al. [98] studied hydrocortisone as a model lipophilic drug and investigated the effects of cosurfactants on the transdermal delivery. Ethyl alcohol, isopropanol, and 1,2-propanediol (propylene glycol) were used as cosurfactants in the microemulsion. The results showed a significantly increased transdermal flux of hydrocortisone from MEs containing these cosurfactants compared to a cosurfactant-free formulation. Thus, the incorporation of cosurfactants not only modifies the physical characteristics of the microemulsion but it also increases the permeation efficacy, supporting the conception of the nanovesicle-driven mechanism of skin penetration [8, 68–70]. Carvalho et al. [97] compared a non-aqueous microemulsion (containing propylene glycol) with an aqueous microemulsion (containing water), and evaluated their ability to improve drug delivery into the skin. The researchers studied the delivery of progesterone (MW=314.5 g/mol, logP=3.87), α- tocopherol (MW=430.7 g/mol, logP=7.96), and lycopene (MW=537, logP=9.16). The results of the skin penetration studies have demonstrated that the penetration enhancement promoted by the aqueous formulation was significant for progesterone and tocopherol, but not for lycopene. These data suggest that skin penetration of highly lipophilic drug is limited by aqueous MEs, and its use by this system may be redundant. Other promising results have been shown for repaglinide. This drug is a quick and short-acting BCS class II drug, and due to its poor water solubility (logP=5.4), and its short half- life (~1 h) (rapid first-pass metabolism), it represents a good candidate for microemulsion systems. Ex vivo permeability study across rat skin showed a 12.3-fold increase in flux with a microemulsion formulation compared to repaglinide suspension in water [1].
SLNs, on the other hand, are entirely different nanocarriers. Their main advantages include the protection of labile substances from chemical degradation, controlled drug release due to the solid state of the lipid matrix, and film formation on the skin that produces an occlusion effect. The occlusion effect leads to a reduced water loss and increased skin hydration, and produces a significant reduction in the total Transepidermal Water Loss (TEWL) [109]. The occlusion effect ability of SLNs (composed of glyceryl dibehenate) was studied using porcine skin. Occlusion Factor (F) was calculated according to Teeranachaideekul et al. [96], where F=0 means no occlusive effect while an F=100 means maximum occlusion. In this study, an occlusion factor of 36% for SLNs was shown. Furthermore, TEWL (recorded with a Tewameter®) showed a 34.3% reduction in TEWL. The studies have confirmed that SLNs, by their affinity to the stratum corneum, form an invisible and occlusive film over the skin that reduces transepidermal water loss and improves skin hydration [96, 115–117]. One more characteristic of SLNs is its lipophilic nature that combined with the high surface area allows longer contact duration of the drug with the skin and results in an efficient drug delivery. However, only thermostable drugs should be chosen for the SLN system due to the high temperatures required to be applied during production [89]. SLNs’ components are also relevant for transdermal absorption as their properties are critical for getting optimal transdermal permeation. Some interesting results have been published by Gaur et al. [110], who studied SLNs loaded with curcumin and obtained high drug permeation through human skin after 24h by including ceramide- 2 in the formulation. The amounts and the proportions of components may also influence the SLN properties as shown by Wake et al. [111], who designed SLNs for transdermal rasagiline mesylate to alleviate the symptoms of Parkinson’s disease. This research has shown that increasing the concentration of stearic acid in the SLNs led to a higher entrapment while increasing the concentration of tween 80 led to a smaller particle size. Unlike liquid oils and microemulsions, SLNs are able to release drugs by a controlled manner, while the slow mobility of the drug molecules is dictated by the diffusion out of the solid lipid. SLNs are usually made of physiological lipids such as fatty acids, steroids, monoglycerides, diglycerides and triglycerides [118], therefore they are considered safe and the danger of acute and chronic toxicity is much lower. However, its cytotoxicity will depend on the compounds that form the lipid matrix. Weyenberg et al. [119] tested a series of positively and negatively surface charged SLNs, demonstrating a cytotoxicity caused by the influence of surfactant excipients. Moreover, significant cytotoxicity was measured when SLNs were coated with the cationic surfactant cetylpyridinium chloride, while formulations containing Lipoid S75 reduced cell viability just slightly.
PNPs are different from MEs and SLNs by their rigidity, which might be critical for transdermal drug delivery. Although polymeric nanoparticles have a potential as transdermal drug carriers, the penetration of nanoparticles into the skin may be significantly lower due to this rigidity when comparing with soft nanoparticles (e.g., liposomes) [120]. It is, therefore, advisable to evaluate the specific PNPs’ properties and to consider a surface modification in order to obtain a significant skin penetration. Marimuthu et al. [121] prepared poly (lactide-co-glycolide) (PLGA) nanoparticles containing glucosamine and modified their outer surface, resulting in a more flexible permeability through the skin lipid membranes compared with uncoated glucosamine nanoparticles. Polymeric nanoparticles are generally prepared by one of three techniques – emulsion/solvent evaporation, nanoprecipitation, and salting out – all of them need a volatile organic solvent in which a hydrophobic drug is dissolved. Thus, PNPs are a useful reservoir for hydrophobic drugs designed for delivering them through the stratum corneum and for releasing these drugs by a controlled manner into the viable skin. Luengo et al. [100] used flufenamic acid as a lipophilic drug model and reported accumulation of the drug in deeper layers of excised human skin after more than 12 h. Transdermal hydrophilic drugs have also been studied using PLGA nanoparticles. Procaine hydrochloride-loaded PLGA nanoparticles, having an average diameter of 150–210 nm, were reported to increase skin accumulation, following by elevated muscle concentrations when using isopropyl myristate as a transdermal enhancer [122].
Perspectives for Transdermal Dosage Forms
The advantages of the dermal and transdermal drug route of administration are the best incentive to keep investing efforts and spending time in its research. The benefits of this route are, just to mention a few, the large surface area of the skin that provides a vast accessible area for drugs, the patient-friendly application that helps to improve the treatment adherence, the avoidance of the gastrointestinal tract environment, the decrease of side effects, as well as reducing multiple dosing frequencies. Due to the low number of drug molecules that are capable of passively crossing the skin barrier, development and application of nanocarriers such as MEs, SLNs, and PNPs are needed to expand the scope of drug compounds that would be quantitatively transported through the skin. More innovative formulation work, pre-clinical experiments, and clinical studies need to be done to establish the safety and efficacy of these nanosystems before introducing into the market. The current reports have demonstrated the advantages of nanometric transdermal formulations; however, solvent-free, safe and non-irritant excipients, time-consuming and inexpensive manufacturing process should be taken into consideration during product development. The number of next- generation therapeutics is expected to increase with new types of drug molecules, like proteins, peptides, and antibodies, even though the future challenge of the pharmaceutical scientists would be tougher and more difficult. The toughest obstacle of all might be the need to hold an active macromolecule into a nanoparticle or a nanodroplet while preserving its activity once it releases into the skin or in the systemic circulation. Altogether, the nanosystems have become successful dosage forms for a variety of dermally and transdermally active principals by modulating drug transfer and serving as nontoxic penetration enhancers.
References
- Shinde, Ujwala A, Sheela H Modani, Kavita H Singh (2018) Design and Development of Repaglinide Microemulsion Gel for Transdermal Delivery. AAPS PharmSciTech 19: 315–25.
- Darreh-Shori, T, V Jelic (2010) Safety and Tolerability of Transdermal and Oral Rivastigmine in Alzheimer’s Disease and Parkinson’s Disease Dementia. Expert Opinion on Drug Safety 9: 167–76.
- Pastore, Michael N, Yogeshvar N Kalia, Michael Horstmann, Michael S Roberts (2015) Transdermal Patches: History, Development and Pharmacology. British journal of pharmacology 172: 2179–209.
- Huang Da, Mi Sun, Yazhong Bu, Fang Luo, Cuiying Lin, et al (2017) Microcapsule-embedded hydrogel patches for ultrasound responsive and enhanced transdermal delivery of diclofenac sodium. J Mater Chem B 5: 8653–8675.
- Tosato MG, JV Maya Giron, AA Martin, V Krishna Tippavajhala, M Fernandez Lorenzo de Mele et al (2018) Comparative Study of Transdermal Drug Delivery Systems of Resveratrol: High Efficiency of Deformable Liposomes. Mater Sci Eng C Mater Biol Appl 90: 356–64.
- Wu, Xiao-mei, Christopher J. Branford-White, Li-min Zhu, Nichoals P. Chatterton, and Deng-guang Yu (2010) Ester Prodrug-Loaded Electrospun Cellulose Acetate Fiber Mats as Transdermal Drug Delivery Systems.” Journal of Materials Science: Materials in Medicine 21: 2403–11.
- Williams, Adrian C, and Brian W Barry (2012) Penetration Enhancers. Advanced Drug Delivery Reviews 64: 128–37.
- Sintov, Amnon C, Shafir Botner (2006) Transdermal Drug Delivery Using Microemulsion and Aqueous Systems: Influence of Skin Storage Conditions on the in Vitro Permeability of Diclofenac from Aqueous Vehicle Systems.” International journal of pharmaceutics 311: 55–62.
- Jain SK, MK Chourasia, R Masuriha, V Soni, A Jain, Nitin K Jain, and Y Gupta. “Solid Lipid Nanoparticles Bearing Flurbiprofen for Transdermal Delivery. Drug delivery 12: 207–15.
- Hatanaka, Tomomi, Toshihiko Kamon, Setsuko Morigaki, Kazunori Katayama, et al (2000) Ion Pair Skin Transport of a Zwitterionic Drug, Cephalexin. Journal of Controlled Release 66: 63–71.
- Paudel, Kalpana S, Mikolaj Milewski, Courtney L Swadley, Nicole K Brogden et al (2010) Challenges and Opportunities in Dermal/Transdermal Delivery. Therapeutic delivery : 109–31.
- Bodkin, J Alexander, and Jay D Amsterdam. “Transdermal Selegiline in Major Depression: A Double- Blind, Placebo-Controlled, Parallel-Group Study in Outpatients. American Journal of Psychiatry 11: 1869–75.
- –Winblad, Bengt, Jeffrey Cummings, Niels Andreasen, George Grossberg, et al (2007) A Six–Month Double–Blind, Randomized, Placebo–Controlled Study of a Transdermal Patch in Alzheimer’s Disease––Rivastigmine Patch Versus Capsule. International Journal of Geriatric Psychiatry: A journal of the psychiatry of late life allied sciences 22: 456–67.
- Chelly, Jacques E, Jeffrey Grass, Timothy W Houseman, Harold Minkowitz, et al (2004) The Safety and Efficacy of a Fentanyl Patient-Controlled Transdermal System for Acute Postoperative Analgesia: A Multicenter, Placebo-Controlled Trial. Anesthesia Analgesia 98: 427–33.
- Xu, Peng-Gang, Xi-Feng Lei, Bao-Di Ren, Shui-Ying Lv, et al (2017) Diclofenac Transdermal Patch Versus the Sustained Release Tablet: A Randomized Clinical Trial in Rheumatoid Arthritic Patients. Tropical Journal of Pharmaceutical Research 16: 477–82.
- Bhaskar, Kesavan, Jayaraman Anbu, Velayutham Ravichandiran, Vobalaboina Venkateswarlu, et al (2009) Lipid Nanoparticles for Transdermal Delivery of Flurbiprofen: Formulation, in Vitro, Ex Vivo and in Vivo Studies. Lipids in health disease 8: 6.
- Harirforoosh, Sam, Waheed Asghar, Fakhreddin Jamali (2014) Adverse Effects of Nonsteroidal Antiinflammatory Drugs: An Update of Gastrointestinal, Cardiovascular and Renal Complications. Journal of Pharmacy Pharmaceutical Sciences 16: 821–47.
- Charoo, Naseem Ahmad, Areeg Anwer Ali Shamsher, Kanchan Kohli, et al (2008) Improvement in Bioavailability of Transdermally Applied Flurbiprofen Using Tulsi (Ocimum Sanctum) and Turpentine Oil. Colloids surfaces B: Biointerfaces 65: 300–07.
- Apfel, Christian C, Kun Zhang, Elizabeth George, Serena Shi, et al (2010) Transdermal Scopolamine for the Prevention of Postoperative Nausea and Vomiting: A Systematic Review and Meta-Analysis. Clinical therapeutics 32: 1987–2002.
- Pergolizzi Jr, Joseph V, Beverly K Philip, John B Leslie, Robert Taylor Jr et al (2012) Perspectives on Transdermal Scopolamine for the Treatment of Postoperative Nausea and Vomiting. Journal of clinical anesthesia 24: 334–45.
- Kaestli, Laure-Zoé, Anne-Florence Wasilewski-Rasca, Pascal Bonnabry, Nicole Vogt-Ferrier (2008) Use of Transdermal Drug Formulations in the Elderly. Drugs aging 25: 269–80.
- Delgado-Charro, M Begona, and Richard H Guy (2014) Effective Use of Transdermal Drug Delivery in Children. Advanced Drug Delivery Reviews 73: 63–82.
- Boateng, Joshua (2017) Drug Delivery Innovations to Address Global Health Challenges for Pediatric and Geriatric Populations (through Improvements in Patient Compliance). Journal of pharmaceutical sciences 106: 3188–98.
- Gnjidic, Danijela, Andy Husband, Adam Todd (2009) Challenges and Innovations of Delivering Medicines to Older Adults. Advanced Drug Delivery Reviews.
- Bos, Jan D, Marcus MHM Meinardi (2000) The 500 Dalton Rule for the Skin Penetration of Chemical Compounds and Drugs. Experimental Dermatology: Viewpoint 9: 165–69.
- Delgado-Charro, M Begona, Richard H Guy (2003) Iontophoresis: Applications in Drug Delivery and Noninvasive Monitoring.” Transdermal Drug Delivery Systems 2: 199–225.
- Prausnitz, Mark R, Samir Mitragotri, Robert Langer (2004) Current Status and Future Potential of Transdermal Drug Delivery. Nature reviews Drug discovery 3: 115.
- Huang, Yaw-Bin, Yi-Hung Tsai, Jui-Sheng Chang, Jian Chen Liu, et al (2002) Effect of Antioxidants and Anti-Irritants on the Stability, Skin Irritation and Penetration Capacity of Captopril Gel. International Journal of Pharmaceutics 241: 345–51.
- Larsen, Rikke H, Flemming Nielsen, Jens A Sørensen, Jesper B Nielsen (2003) Dermal Penetration of Fentanyl: Inter-and Intraindividual Variations. Pharmacology toxicology 93: 244–48.
- Manevski, Nenad, Piet Swart, Kamal Kumar Balavenkatraman, Barbara Bertschi, et al (2015) Phase Ii Metabolism in Human Skin: Skin Explants Show Full Coverage for Glucuronidation, Sulfation, N-Acetylation, Catechol Methylation, and Glutathione Conjugation. Drug Metabolism Disposition 43: 126–39.
- Wilkinson, Simon C, Faith M Williams (2007) Cutaneous Metabolism. Dermal absorption toxicity assessment : 89–115.
- Jacques C, E Perdu, EL Jamin, JP Cravedi, A Mavon, et al (2014) Effect of Skin Metabolism on Dermal Delivery of Testosterone: Qualitative Assessment Using a New Short-Term Skin Model. Skin pharmacology physiology 27 : 188–88.
- Fang, Jia-You, Yann-Lii Leu (2006) Prodrug Strategy for Enhancing Drug Delivery via Skin. Current drug discovery technologies 3: 211–24.
- Doh, Hea-Jeong, Won-Jea Cho, Chul-Soon Yong, Han-Gon Choi, et al (2003) Synthesis and Evaluation of Ketorolac Ester Prodrugs for Transdermal Delivery. Journal of pharmaceutical sciences 92: 1008–17.
- Walters, Kenneth A, Michael S Roberts (2002) The structure and function of skin. In Dermatological and Transdermal Formulations, 1st ed.: Walters, KA, Eds.; Marcel Dekker, Inc., New York, USA, Pg No: 1–39.
- Tobin, Desmond J (2006) Biochemistry of Human Skin—Our Brain on the Outside. Chemical Society Reviews 35: 52–67.
- Illel, Brigitte, Hans Schaefer, Jacques Wepierre, Olivier Doucet (1991) Follicles Play an Important Role in Percutaneous Absorption. Journal of pharmaceutical sciences 80: 424–27.
- Pohlmann, Adriana R, Silvia S Guterres, Tatiele Katzer, Renata Vidor Contri (2016) Drug Transport across Skin. In Drug Delivery across Physiological Barriers 143–66.
- Ghafourian, Taravat, Parinaz Zandasrar, Hamed Hamishekar, Ali Nokhodchi (2004) The Effect of Penetration Enhancers on Drug Delivery through Skin: A Qsar Study. Journal of Controlled Release 99: 113–25.
- Chen, Longjian, Guoping Lian, Lujia Han (2010) Modeling Transdermal Permeation. Part I. Predicting Skin Permeability of both Hydrophobic and Hydrophilic Solutes. AIChE journal 56: 1136–46.
- Ng, Keng Wooi, Wing Man Lau (2015) Skin Deep: The Basics of Human Skin Structure and Drug Penetration. In Percutaneous Penetration Enhancers Chemical Methods in Penetration Enhancement: Drug Manipulation Strategies and Vehicle Effects, edited by Nina Dragicevic and Howard I. Maibach, 3–11. Berlin, Heidelberg: Springer Berlin Heidelberg 2015.
- Hueber, Frédérique, Jacques Wepierre, Hans Schaefer (1992) Role of Transepidermal and Transfollicular Routes in Percutaneous Absorption of Hydrocortisone and Testosterone: In Vivo Study in the Hairless Rat. Skin pharmacology physiology 5: 99–107.
- Tampucci S, Burgalassi S, Chetoni P, Lenzi C, Pirone A, et al (2014) Topical formulations containing finasteride. Part II: determination of finasteride penetration into hair follicles using the differential stripping technique. J Pharm Sci 103: 2323–9.
- Alvarez-Román, Rocio, Aarti Naik, YN Kalia, Richard H Guy, H Fessi (2004) Skin Penetration and Distribution of Polymeric Nanoparticles. Journal of Controlled Release 99: 53–62.
- Raber AS, Mittal A, Schaefer J, Bakowsky U, Reichrath J, et al (2014) Quantification of nanoparticle uptake into hair follicles in pig ear and human forearm. J Control Release 179: 25–32.
- Patzelt, Alexa, Heike Richter, Lars Dähne, Peter Walden, et al (2011) Influence of the Vehicle on the Penetration of Particles into Hair Follicles. Pharmaceutics 3: 307–14.
- Grice, Jeffrey E, Hamid R Moghimi, Elizabeth Ryan, Qian Zhang,et al (2017) Non-Formulation Parameters That Affect Penetrant-Skin-Vehicle Interactions and Percutaneous Absorption. In Percutaneous Penetration Enhancers Drug Penetration into/through the Skin 45–75.
- Wang, Yiping, Rashmi Thakur, Qiuxi Fan, Bozena Michniak (2005) Transdermal Iontophoresis: Combination Strategies to Improve Transdermal Iontophoretic Drug Delivery. European Journal of Pharmaceutics Biopharmaceutics 60: 179–91.
- Prausnitz, Mark R, Vanu G Bose, Robert Langer, James C Weaver (1993) Electroporation of Mammalian Skin: A Mechanism to Enhance Transdermal Drug Delivery.” Proceedings of the National Academy of Sciences 90: 10504–08.
- Mitragotri, Samir, Daniel Blankschtein, Robert Langer (1996) Transdermal Drug Delivery Using Low- Frequency Sonophoresis. Pharm Res 13: 411–20.
- Yu, Jing, Dhaval R Kalaria, and Yogeshvar N Kalia (2011) Erbium: Yag Fractional Laser Ablation for the Percutaneous Delivery of Intact Functional Therapeutic Antibodies. Journal of controlled release 156: 53–59.
- Martanto, Wijaya, Shawn P Davis, Nicholas R Holiday, Jenny Wang, et al (2004) Transdermal Delivery of Insulin Using Microneedles in Vivo. Pharm Res 21: 947–52.
- Funke, Adrian P, Roman Schiller, Hans W Motzkus, Clemens Günther, (2002) Transdermal Delivery of Highly Lipophilic Drugs: In Vitro Fluxes of Antiestrogens, Permeation Enhancers, and Solvents from Liquid Formulations. Pharm Res 19: 661–68.
- Lu, Guang Wei, Ping Gao (2010) Chapter 3 – Emulsions and Microemulsions for Topical and Transdermal Drug Delivery. In Handbook of Non-Invasive Drug Delivery Systems, edited by Vitthal S. Kulkarni, 59–94. Boston: William Andrew Publishing 10003–4.
- Baroli, Bianca, M Arturo López-Quintela, M Begoña Delgado-Charro, Anna M Fadda, et al (2000) Microemulsions for Topical Delivery of 8-Methoxsalen. Journal of Controlled Release 69: 209–18.
- Chen, Huabing, Xueling Chang, Ting Weng, Xiaozhi Zhao (2004) A Study of Microemulsion Systems for Transdermal Delivery of Triptolide. Journal of Controlled Release 98: 427–36.
- Biruss, Babette, Hanspeter Kählig, Claudia Valenta (2007) Evaluation of an Eucalyptus Oil Containing Topical Drug Delivery System for Selected Steroid Hormones. International Journal of Pharmaceutics 328: 142–51.
- Lopes, Luciana B, Hillary VanDeWall, Hsin T Li, Vijay Venugopal, Hsin K Li, et al (2010) Topical Delivery of Lycopene Using Microemulsions: Enhanced Skin Penetration and Tissue Antioxidant Activity. Journal of pharmaceutical sciences 99: 1346–57.
- Lopes, Luciana (2014) Overcoming the Cutaneous Barrier with Microemulsions. Pharmaceutics 6: 52–77.
- Gannu, Ramesh, Chinna Reddy Palem, Vamshi Vishnu Yamsani, Shravan Kumar Yamsani, Madhusudan Rao Yamsani (2010) Enhanced Bioavailability of Lacidipine via Microemulsion Based Transdermal Gels: Formulation Optimization, Ex Vivo and in Vivo Characterization. International Journal of Pharmaceutics 388: 231–41.
- Said, Mayada, Ibrahim Elsayed, Ahmed A Aboelwafa, Ahmed H Elshafeey (2017) Transdermal Agomelatine Microemulsion Gel: Pyramidal Screening, Statistical Optimization and in Vivo Bioavailability. Drug delivery 24: 1159–69.
- Shukla, Tripti, Neeraj Upmanyu, Mukta Agrawal, Swarnlata Saraf,et al (2018) Biomedical Applications of Microemulsion through Dermal and Transdermal Route. Biomedicine Pharmacotherapy 108: 1477–94.
- Ghosh, Bijaya, L Harivardhan Reddy (2001) Effect of Physicochemical Parameters on Skin Permeability of Antihypertensive. Indian Journal of Experimental Biology 39: 710 – 14.
- Hathout, Rania M, Samar Mansour, Nahed D Mortada, Ahmed S Geneidi, Richard H Guy (2010) Uptake of Microemulsion Components into the Stratum Corneum and Their Molecular Effects on Skin Barrier Function. Mol Pharm 7: 1266–73.
- Naik, Aarti, Louk ARM Pechtold, Russell O Potts, Richard H Guy (1995) Mechanism of Oleic Acid- Induced Skin Penetration Enhancement in Vivo in Humans. Journal of Controlled Release 37: 299–306.
- El Khayat, Noha W, Ahmed A Donia, Omer Y Mady, Gamal M El Maghraby (2018) Optimization of Eugenol Microemulsion for Transdermal Delivery of Indomethacin. Journal of Drug Delivery Science Technology 48: 311–18.
- Carvalho, André Luis Menezes, José Alexsandro Silva, Ana Amélia Moreira Lira, Ellen Denise Prado Almeida, (2018) Third-Generation Transdermal Delivery Systems Containing Zidovudine: Effect of the Combination of Different Chemical Enhancers and a Microemulsion System. AAPS PharmSciTech 19: 3219–27.
- Sintov AC, L Shapiro (2004) New Microemulsion Vehicle Facilitates Percutaneous Penetration in Vitro and Cutaneous Drug Bioavailability in Vivo. Journal of Controlled Release 95: 173–83.
- Sintov AC, I Greenberg (2014) Comparative Percutaneous Permeation Study Using Caffeine-Loaded Microemulsion Showing Low Reliability of the Frozen/Thawed Skin Models. International Journal of Pharmaceutics 471: 516–24.
- Sintov AC (2015) Transdermal Delivery of Curcumin Via Microemulsion. International Journal of Pharmaceutics 481: 97–103.
- Budhian, Avinash, Steven J Siegel, Karen I Winey (2005) Production of Haloperidol-Loaded Plga Nanoparticles for Extended Controlled Drug Release of Haloperidol. Journal of microencapsulation 22: 773–785.
- Mu, Liang, SSFeng (2003). A Novel Controlled Release Formulation for the Anti cancer Drug Paclitaxel (Taxol®): Plga Nanoparticles Containing Vitamin E Tpgs. Journal of Controlled Release 86: 33–48.
- Lale, Shantanu V, AthulyaAravind, D Sakthi Kumar, VeenaKoul (2014) As1411 Aptamer and Folic Acid Functionalized Ph-Responsive Atrp Fabricated Ppegma–Pcl–Ppegma Polymeric Nanoparticles for Targeted Drug Delivery in Cancer Therapy. Biomacromolecules 15: 1737–1752.
- Das, Pradip, NikhilRJana (2015). Dopamine Functionalized Polymeric Nanoparticle for Targeted Drug Delivery. RSC Advances 5: 33586–33594.
- Al-Otoum, R, SRA bulateefeh, and MOTaha (2014). Preparation of Novel Ionotropically Crosslinked Beads Based on Alginate-Terephthalic Acid Composites as Potential Controlled Release Matrices. Die Pharmazie-An International Journal of Pharmaceutical Sciences 69: 10–18.
- Ezpeleta, Isabel, Juan M Irache, Serge Stainmesse, Christiane Chabenat,et al (1996) Gliadin Nanoparticles for the Controlled Release of All- Trans-Retinoic Acid. International Journal of Pharmaceutics 131: 191–200.
- Montero, Nuria, Elena Pérez, Marta Benito, César Teijón, et al (2019) Biocompatibility Studies of Intravenously Administered Ionic-Crosslinked Chitosan-Bsa Nanoparticles as Vehicles for Antitumour Drugs. International Journal of Pharmaceutics 554: 337–351.
- Jiang, Xiaoyu, Hongting Pu, Peng Wang (2011) Polymer Nanoparticles Via Intramolecular Crosslinking of Sulfonyl Azide Functionalized Polymers. Polymer 52: 3597–602.
- Sheng, Yan, Changsheng Liu, Yuan Yuan, Xinyi Tao, (2009) Long-Circulating Polymeric Nanoparticles Bearing a Combinatorial Coating of Peg and Water-Soluble Chitosan. Biomaterials 30: 2340–48.
- Chauvierre, Cédric, Denis Labarre, Patrick Couvreur, and Christine Vauthier (2003) Novel Polysaccharide- Decorated Poly (Isobutyl Cyanoacrylate) Nanoparticles. Pharm Res 20: 1786–93.
- Massella, Daniele, Federica Leone, Roberta Peila, Antonello Barresi, and Ada Ferri (2018) Functionalization of Cotton Fabrics with Polycaprolactone Nanoparticles for Transdermal Release of Melatonin. Journal of functional biomaterials 9: 1.
- Abnoos, Marzieh, Mojdeh Mohseni, Seyed Ali Javad Mousavi, Khadijeh Ashtari, et al (2018) Chitosan-Alginate Nano-Carrier for Transdermal Delivery of Pirfenidone in Idiopathic Pulmonary Fibrosis. International journal of biological macromolecules.
- Garces, A, MH Amaral, JM Sousa Lobo, AC Silva (2018) Formulations Based on Solid Lipid Nanoparticles (Sln) and Nanostructured Lipid Carriers (Nlc) for Cutaneous Use: A Review. European Journal of Pharmaceutical Sciences 112: 159–67.
- Müller, Rainer H, Dörte Rühl, Stephan Runge, Kai Schulze-Forster, Wolfgang Mehnert (1997) Cytotoxicity of Solid Lipid Nanoparticles as a Function of the Lipid Matrix and the Surfactant. Pharm Res 14: 458–62.
- Shegokar R, KK Singh, RH Müller (2011) Production & Stability of Stavudine Solid Lipid Nanoparticles—from Lab to Industrial Scale. International Journal of Pharmaceutics 416: 461–70.
- Guo, Dawei, Dandan Dou, Xinyu Li, Qian Zhang (2018) Ivermection-Loaded Solid Lipid Nanoparticles: Preparation, Characterisation, Stability and Transdermal Behaviour. Artificial cells, nanomedicine, biotechnology 46: 255–62.
- Mehnert, Wolfgang, Karsten Mäder (2012) Solid Lipid Nanoparticles: Production, Characterization and Applications. Advanced Drug Delivery Reviews 64: 83–101.
- Bhalekar, Mangesh, Prashant Upadhaya, and Ashwini Madgulkar (2017) Formulation and Characterization of Solid Lipid Nanoparticles for an Anti-Retroviral Drug Darunavir. Applied Nanoscience 7: 47–57.
- Ramteke, KH, SA Joshi, SN Dhole (2012) Solid Lipid Nanoparticle: A Review. IOSR journal of pharmacy 2 : 34–44.
- Gallarate, Marina, Luigi Battaglia, Elena Peira, Michele Trotta (2011) Peptide-Loaded Solid Lipid Nanoparticles Prepared through Coacervation Technique. International Journal of Chemical Engineering.
- Hamishehkar, Hamed, Saeideh Same, Khosro Adibkia, Kamyar Zarza, et al (2015) A Comparative Histological Study on the Skin Occlusion Performance of a Cream Made of Solid Lipid Nanoparticles and Vaseline. Research in pharmaceutical sciences 10: 378.
- Gönüllü, Ümit, Melike Üner, Gülgün Yener, Ecem Fatma Karaman, Zeynep Aydoğmuş (2015) Formulation and Characterization of Solid Lipid Nanoparticles, Nanostructured Lipid Carriers and Nanoemulsion of Lornoxicam for Transdermal Delivery. Acta Pharmaceutica 65: 1–13.
- Ravi, G, Gupta, VN and Balamuralidhara,V (2018) Rivastigmine Tartrate Solid Lipid Nanoparticles Loaded Transdermal Film: An in Vivo Study. Research Journal of Pharmacy Technology 11: 227–227.
- Mueller, RH, Radtke M, Wissing (2002) Nanostructured lipid matrices for improved microencapsulation of drugs. International Journal of Pharmacetics 242: 121–128.
- Chandrashekar, NS, and RH Shobha Rani (2008) Physicochemical and Pharmacokinetic Parameters in Drug Selection and Loading for Transdermal Drug Delivery. Indian journal of pharmaceutical sciences 70: 94.
- Teeranachaideekul V, Boonme, P, Souto, EB, Mueller, RH, Junyaprasert, VB (2008) Influence of oil content on physiochemical properties and skin distribution of Nile red-loaded NLC. Journal of Controlled Release 128: 134–141.
- Carvalho, Vanessa F, Debora P de Lemos, Camila S Vieira, Amanda Migotto, Luciana B Lopes (2017) Potential of Non-Aqueous Microemulsions to Improve the Delivery of Lipophilic Drugs to the Skin. AAPS PharmSciTech 18 : 1739–49.
- El Maghraby, Gamal M (2008) Transdermal Delivery of Hydrocortisone from Eucalyptus Oil Microemulsion: Effects of Cosurfactants. International Journal of Pharmaceutics 355: 285–92.
- Sood, Jatin, Bharti Sapra, Ashok K Tiwary (2017) Microemulsion Transdermal Formulation for Simultaneous Delivery of Valsartan and Nifedipine: Formulation by Design. AAPS PharmSciTech 18: 1901–16.
- Soliman, Sara M, NS Abdel Malak, Omaima N El-Gazayerly, AA Abdel Rehim (2010) Formulation of Microemulsion Gel Systems for Transdermal Delivery of Celecoxib: In Vitro Permeation, Anti- Inflammatory Activity and Skin Irritation Tests. Drug Discov Ther 4: 459–71.
- Dave, Kaushalkumar, Fahd M Alsharif, Saiful Islam, Chandradhar Dwivedi, et al (2017) Chemoprevention of Breast Cancer by Transdermal Delivery of Α-Santalol through Breast Skin and Mammary Papilla (Nipple). Pharm Res 34: 1897–907.
- Jain, Sanyog, Ankit Mittal, Amit K Jain (2011) Enhanced Topical Delivery of Cyclosporin-a Using Plga Nanoparticles as Carrier. Current Nanoscience 7: 524–30.
- El-Gogary, Riham I, Noelia Rubio, Julie Tzu-Wen Wang, Wafa’T Al-Jamal, et al (2014) Polyethylene Glycol Conjugated Polymeric Nanocapsules for Targeted Delivery of Quercetin to Folate-Expressing Cancer Cells in Vitro and in Vivo. Acs Nano 8: 1384–401.
- Dias, Sávia Francisca Lopes, Silvania Siqueira Nogueira, Flaviane de França Dourado, Maria Adelaide Guimarães, et al (2016) Acetylated Cashew Gum-Based Nanoparticles for Transdermal Delivery of Diclofenac Diethyl Amine. Carbohydrate polymers 143: 254–261,
- Luengo J, B Weiss, M Schneider, A Ehlers, F Stracke, et al (2006) Influence of Nanoencapsulation on Human Skin Transport of Flufenamic Acid. Skin pharmacology physiology 19: 190–97.
- Al-Kassas, Raida, Jingyuan Wen, Angel En-Miao Cheng, Amy Moon-Jung Kim, et al (2016) Transdermal Delivery of Propranolol Hydrochloride through Chitosan Nanoparticles Dispersed in Mucoadhesive Gel. Carbohydrate polymers 153: 176–86.
- Hafner, Anita, Jasmina Lovrić, Ivan Pepić, Jelena Filipović-Grčić (2011) Lecithin/Chitosan Nanoparticles for Transdermal Delivery of Melatonin. Journal of microencapsulation 28: 807–15.
- Sutthanut, Khaetthareeya, Xiuling Lu, Michael Jay, Bungorn Sripanidkulchai (2009) Solid Lipid Nanoparticles for Topical Administration of Kaempferia Parviflora Extracts. Journal of biomedical nanotechnology 5: 224–32.
- Kurakula, Mallesh, Osama AA Ahmed, Usama A Fahmy, Tarek A Ahmed (2016) Solid Lipid Nanoparticles for Transdermal Delivery of Avanafil: Optimization, Formulation, in-Vitro and Ex-Vivo Studies.” Journal of liposome research 26: 288–96.
- Gaur, Praveen Kumar, Shikha Mishra, Anurag Verma, Navneet Verma (2016) Ceramide–Palmitic Acid Complex Based Curcumin Solid Lipid Nanoparticles for Transdermal Delivery: Pharmacokinetic and Pharmacodynamic Study. Journal of Experimental Nanoscience 11: 38–53.
- Wake, Prashant S, and MD Kshirsagar (2017) Compatibility Study in-Vitro Drug Release Study of Solid Lipid Nanoparticle Based Transdermal Drug Delivery System for Rasagiline Mesylate. Asian Journal of Research in Pharmaceutical Science 7: 92–96.
- Puglia, Carmelo, Alessia Offerta, Giorgia Giusy Tirendi, Maria Stella Tarico, et al (2016) Design of Solid Lipid Nanoparticles for Caffeine Topical Administration. Drug delivery 23: 36–40.
- Boltri, L, S Morel, M Trotta, and MR Gasco (1994) In Vitro Transdermal Permeation of Nifedipine from Thickened Microemulsions. Journal de pharmacie de Belgique 49: 315–20.
- Changez, Mohammad, Jagdish Chander, Amit Kumar Dinda (2006) Transdermal Permeation of Tetracaine Hydrochloride by Lecithin Microemulsion: In Vivo.” Colloids surfaces B: Biointerfaces 48: 58–66.
- Wissing, S, A Lippacher, and R Müller (2001) Investigations on the Occlusive Properties of Solid Lipid Nanoparticles (SLN). Journal of cosmetic science 52: 313–24,
- Müller, Rainer H, Magdalene Radtke, Sylvia A Wissing (2002) Solid Lipid Nanoparticles (SLN) and Nanostructured Lipid Carriers (NLC) in Cosmetic and Dermatological Preparations. Advanced Drug Delivery Reviews 54: S131-S55.
- Müller, RH, RD Petersen, A Hommoss, J Pardeike (2007) Nanostructured Lipid Carriers (NLC) in Cosmetic Dermal Products. Advanced Drug Delivery Reviews 59: 522–30.
- Shidhaye, SS, Reshma Vaidya, Sagar Sutar, Arati Patwardhan, VJ Kadam (2008) Solid Lipid Nanoparticles and Nanostructured Lipid Carriers-Innovative Generations of Solid Lipid Carriers. Current drug delivery 5: 324–31.
- Weyenberg, Wim, Plamen Filev, Dave Van den Plas, Jo Vandervoort et al (2007) Cytotoxicity of Submicron Emulsions and Solid Lipid Nanoparticles for Dermal Application. International Journal of Pharmaceutics 337: 291–98.
- Lopez, Renata FV, Jennifer E Seto, Daniel Blankschtein, Robert Langer (2011) Enhancing the Transdermal Delivery of Rigid Nanoparticles Using the Simultaneous Application of Ultrasound and Sodium Lauryl Sulfate. Biomaterials 32: 933–41.
- Marimuthu, Mohana, Devasier Bennet, Sanghyo Kim (2013) Self-Assembled Nanoparticles of Plga- Conjugated Glucosamine as a Sustained Transdermal Drug Delivery Vehicle. Polymer journal 45: 202.
- Takeuchi, Issei, Keishiro Tomoda, Megumi Koji, Kimiko Makino (2017) Hydrophilic Drug-Loaded PLGA Nanoparticles for Transdermal Delivery. Colloid Polymer Science 295: 977–83.