DOI: 10.31038/JMG.2020314
Abstract
Fabry Disease (FD) is an X-linked disease caused by variants at GLA gene, producing deficient activity of α-galactosidase. An enzyme, leading to progressive lysosomal accumulation of globotriaosylceramide (GL3) and globotriaosylsphingosine (Lyso-Gb3) in cells. More than 900 GLA variants have been associated with FD, some of them with uncertain pathogenicity. Plasma LysoGb3 is a useful diagnostic tool and considered a potential indication for early Enzyme Replacement Therapy (ERT). However, in pediatric population, the genotype and biomarkers measurement are often not enough to define phenotype and to decide the timely initiation of ERT. We report a novel missense hemizygous mutation, c.805G>C (p.Val269Leu) which caused Fabry nephropathy in a 2 year old male with normal biomarkers.
Introduction
Fabry Disease (FD) is an inherited, genetic condition caused by defects in the GLA gene that are carried by the X-chromosome and thus can be transmitted by the mother and father. Defects in the GLA gene cause deficient or absent lysosomal α-Gal An activity and consequently accumulation of glycosphingolipids mainly GL-3. The progressive GL-3 accumulation in tissues results in cellular and organ damage across the body [1].
More than 900 GLA variants have been reported to date, certain GLA mutations are associated with classic Fabry disease and others with later-onset phenotype [2]. However there is often not enough evidence across individuals for full phenotypic characterization, especially in pediatric population [3].
Measurement of biomarkers like Lyso-Gb3 in plasma has become appreciated as a useful diagnostic tool and considered useful as potential indication for early Enzyme Replacement Therapy (ERT) [4]. However in pediatric population the genotype and biomarkers measurement are often not enough to define phenotype and to decide the timely initiation of ERT. [5].
Clinical Case
We report a new GLA variation in 2-year-old male who was born in Mexico City. His 28-year-old uncle with chronic renal insufficiency was diagnosed with Fabry disease, his 30-year-old mother was an asymptomatic carrier. (Figure 1)
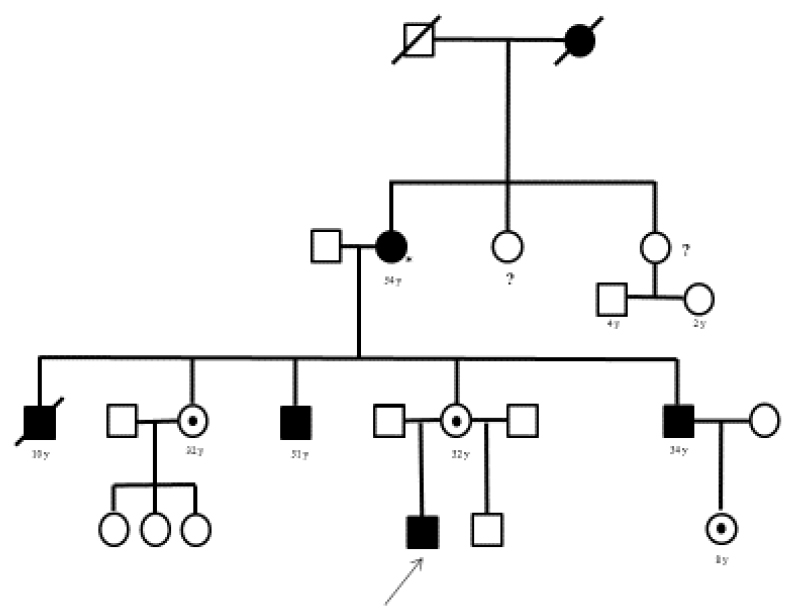
Figure 1. Genographics
On clinical assessment the patient complained of acroparesthesia in the hands and the feet and experienced substantial irritability during the episodes of pain. The physical examination was unremarkable, normal weight and height for age and normal developmental milestones. The enzymatic activity assay in dry blood spot revealed reduced α-galactosidase. An enzymatic activity (0.39nmol/ml/hr; normal range 1.04–5.9 nmol/ml/hr).
We genetically analyzed all coding parts of the GLA gene via PCR and direct sequencing, Molecular analysis revealed that the patient was hemizygous for a missense mutation of exon 6 of the GLA gene c.805G>C (p.Val269Leu). The mutation was previously not described and the p.Val269Leu mutation to our knowledge was detected only in our patient and his family (mother and uncle).
A thorough multisystemic assessment was performed to search for other clinical features of Fabry disease, but no gastrointestinal, cutaneous, ocular, audiologic, cardiologic, neurologic, respiratory or renal involvement was found, only with no significant proteinuria. (Table 1)
Table 1. Clinical Features.
|
Clinical features of patient with V269L mutation |
|
|
Age |
2 years |
|
Gender |
Male |
|
α-galactosidase A (normal 1.04–5.9 nmol/ml/hr) |
0.39 |
|
Lyso Gb3 (<5 ng/ml) |
BQL |
|
Plasma GL-3 (<7 µg/mL) |
3.8 |
|
Urine GL-3 (<7 µg/mL) |
BQL |
|
Symptoms |
|
|
Hypohidrosis |
No |
|
Angiokeratoma |
No |
|
Neuropathic pain |
Yes |
|
Gastrointestinal |
No |
|
Dyspnea |
No |
|
Cardiac |
|
|
EKG |
Normal |
|
IVSD(mm) |
4 |
|
LV mass (g/m2) |
64 |
|
Renal |
|
|
eGFR(Schwartz ml/min) |
115 |
|
Proteinuria (g/24h) |
No-significant |
|
Serum creatinine (mg/dl) |
0.36 |
|
Neurological |
|
|
MRI brain |
Normal |
|
Neuropathy |
Temperature analgesia |
BQL * Bellow quantification levels
The patient’s Lyso-Gb3 plasma level was normal (below quantification levels) measured by tandem mass spectrometer. Moreover, solid-phase chromatography demonstrated that the patient’s Plasma GL-3 level was normal and liquid chromatography of urinary GL-3 level was within normal range. We continued with outpatient follow-up visits for a year and because of the aggressive phenotype of the other members of the family and the presence of acroparesthesia, we decided to perform a kidney biopsy that showed significant GL3 accumulation in several types of kidney cells; GL3 inclusions were present in glomerular endothelial cells, tubular epithelial cells and mesangial cell inclusions were also found. (Figure 2–3)
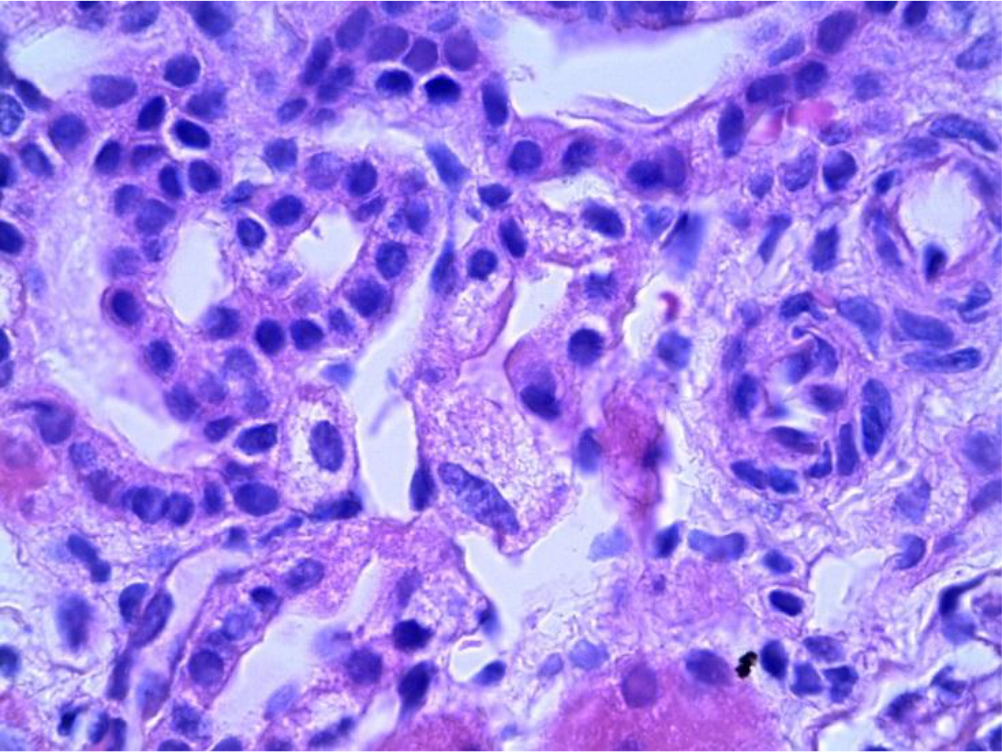
Figure 2. Distal tubules showing abundant inclusions with cytoplasmic expansion (PAS 40X).
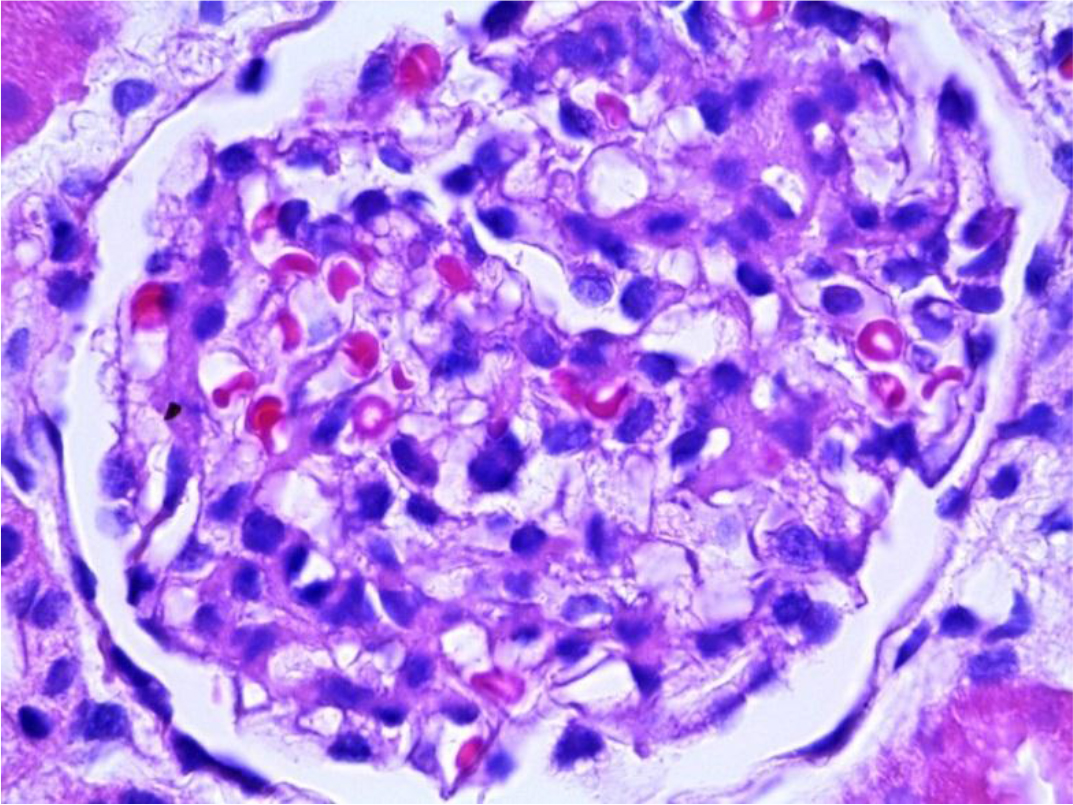
Figure 3. Glomerulus with epithelial cells showing cytoplasmic expansion that becomes vacuolated (PAS 40X).
Electron microscopy showed numerous electron-dense multi-lamellar inclusions in the epithelial cytoplasm with podocyte effacement, typical of Fabry disease. (Figure 4)
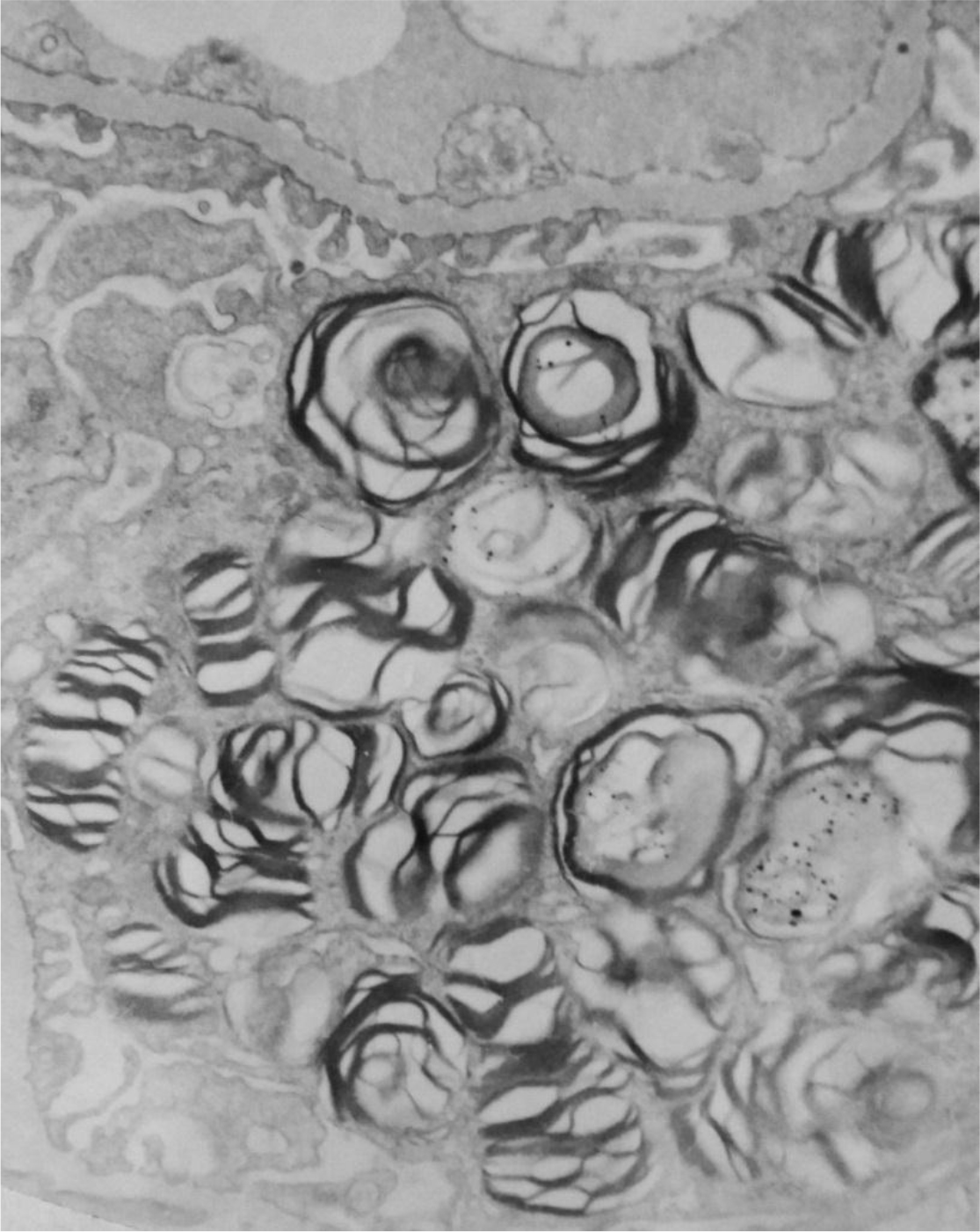
Figure 4. Electronic microscopy showing podocytes with myelin figures and zebra bodies in cytoplasm (4,500x).
Enzyme replacement therapy with agalsidase beta was initiated at 1 mg/kg every other week; during the course of treatment the patient presented a diminished frequency of acroparesthesia.
Discussion
To our knowledge this is the first report of pathogenicity of the p.Val269Leu variant. Three other pathogenic missense mutations have been reported, affecting the same amino acid: p.Val269Met [6], p.Val269Ala [7] and p.Val269Gly, (8) one variant classified as with unknown significance, though without any clinical condition provided (p.Val269Glu)[9], all pointing out a potentially putative role of this amino acid in the protein.
The p.Val269Met mutation was first found in a male patient with classic FD phenotype, together with the p. Asp264Tyr mutation [6]. In vitro analyses demonstrated that while the p.Val269Met mutation led to an approximately 10 per cent of expressed wild-type enzymatic activity, together with the p.D264Y variant, it led to a non-detectable activity in plasma and leukocytes[6]. The p.Val269Ala and p.Val269Gly variants were also detected in patients with classical FD [7–8], and the deleterious effect of the mutations have been confirmed by in vivo measurements of biomarker levels.
Since their first reports, an international reference laboratory confirmed the p.Val269Met variant 65 patients in total (heterozygous/homozygous/hemizygous: 44/0/21, resp.), while p.Val269Ala was confirmed in 4 patients (3/0/1) and p.Val269Gly in 5 patients (2/0/3).
In this case the GLA variant p.Val269Leu demonstrated to be pathogenic in a very young patient with organ tissue damage (kidney and skin biopsy) despite normal biomarkers (lysoGb3, urine GL3 and plasma GL3) highlighting the importance of histologic assessment in children to timely decide initiation of ERT.
Conclusion
In conclusion, we identified a novel missense mutation in GLA, p.Val269Leu, in a 2-year-old male with classic Fabry disease diagnosed with kidney and skin biopsy despite normal biomarkers. The histologic findings provided organ damage information that helped us decide the initiation of ERT. The identification of this novel mutation will enable clinicians and genetic counselors to have better understanding of Fabry disease.
The issue of timely initiation of ERT in childhood is an increasingly important debate, although lysoGb3 is a useful marker, histologic assessment including kidney and skin biopsy may provide more definitive information about organ injury related to FD and better inform treatment decisions, especially when the especially when the GLA variants are novel or variants of uncertain significance.
References
- Ortiz A, Germain DP, Desnick RJ, Politei J, Mauer M et al. (2018) Fabry disease revisited: Management and treatment recommendations for adult patients. Molecular genetics and metabolism 123: 416–427. [Crossref]
- http://www.hgmd,cf,ac,uk/ac/gene.php?gene=GLA
- Oliveira, JP, Ferreira, SI (2019) Multiple phenotypic domains of Fabry disease and their relevance for establishing genotype-phenotype correlations. The application of clinical genetics 12: 35–50. [Crossref]
- Duro G, Zizzo C, Cammarata G, Burlina A, Burlina A (2018). Mutations in the GLA Gene and LysoGb3: Is It Really Anderson-Fabry Disease? International journal of molecular sciences.
- Politei J, Alberton V, Amoreo O, Antongiovanni N, Arán MN et al. (2018). Clinical parameters, LysoGb3, podocyturia, and kidney biopsy in children with Fabry disease: is a correlation possible? Pediatric Nephrology 33: 2095–2101. [Crossref]
- Shabbeer J, Yasuda M, Benson SD, Desnick RJ (2005) Fabry disease: Identification of 50 novel α-galactosidase A mutations causing the classic phenotype and three-dimensional structural analysis of 29 missense mutations. Human Genomics 2: 297–309. [Crossref]
- Davies JP, Winchester BG, Malcolm S (1993) Mutation analysis in patients with the typical form of Anderson-Fabry disease. Human molecular genetics 2: 1051–1053. [Crossref]
- Lukas J, Scalia S, Eichler S, Pockrandt AM, Dehn N et al. (2016). Functional and Clinical Consequences of Novel α-Galactosidase A Mutations in Fabry Disease. Human mutation 37: 43–51. [Crossref]
- https://www.egl-eurofins.com/emvclass/emvclass.php?approved_symbol=GLA