DOI: 10.31038/CST.2020523
Abstract
ACBD3 breast cancer research to date reveals that overexpression at mRNA and protein level is near universal in breast tumour tissue and that high ACBD3 expression is associated with worse patient prognosis. ACBD3 has been shown to have an important role in specifying cell fate and maintaining stem cell pools in neurological development and deletion of ACBD3 in human cell lines prevents cell division. Combined with observations that β-catenin expression and activity is increased when ACBD3 is overexpressed it has been hypothesised that ACBD3 promotes breast cancer by increasing Wnt signalling. This may only be one aspect of ACBD3’s effects as its expression and localisation regulatessteroidogenesis, calcium mediated redox stress and inflammation, glucose import and PI(4)P production which are all intrinsically linked to breast cancer dynamics. Given the wide scope for a role of ACBD3 in breast cancer, we explore its interactors and the implications of preventing these interactions.
Keywords
ACBD3, Breast cancer, Chromosome 1, Golgi, NUMB, PI4Kβ, Phosphatidylinositol, Protein kinase A, Steroidogenesis, Wnt signalling, 1q
Introduction
Although breast cancer incidence has increased in recent years, largely due to improved diagnostic techniques, greater awareness and the introduction of national screening programmes, mortality rates are declining as result of earlier detection and improved treatment regimes. Despite this, treating advanced disease remains difficult and there is a need to identify new therapeutic targets. Proteins encoded by the q-arm of chromosome 1 are of particular interest as regions of 1q are frequently amplified and overexpressed in breast cancer leading to the hypothesis that 1q is important in disease development and progression [1-3]. The frequency at which regions of arm 1q were amplified was investigated and the 1q42.12 locus was found to be amplified in both breast cancer cell lines and primary tumours with a number of genes in or near this region also being over expressed [4]. Of emerging interest is ACBD3, a Golgi protein with multiple functions, only recently linked to breast cancer [5].
ACBD3 was discovered as an interactor of GOLGB1 and named GCP60, and independently discovered as aninteractor of the mitochondrial translocator protein TSPO and protein kinase A and named PAP7 [6,7]. Having found that each of these names were describing one aspect of a diverse adapter protein it was renamed by the HUGO gene nomenclature committee in 2004 as Acetyl CoA Binding Domain containing protein 3, or ACBD3, reflecting its functional groups and protein family rather than any particular role, of which there are many (www.genenames.org). In addition to the acyl CoA binding domain at its N-terminus, ACBD3 contains a Golgi dynamics (GOLD) and a glutamine rich Q domain as well as a proline rich region (Figure 1) [8]. The GOLD domain is found in Golgi and lipid trafficking proteins and makes up the C-terminus of ACBD3 (aa384-526). It is a β-strandrich domain and is responsible for ACBD3 localization to the Golgi via direct interaction with GOLGB1 [6]. ACBD3 is a largely unstructured or loosely structured protein, as many linkers are, and of all the recognisable domains only the GOLD domain structure has been solved by X-ray crystallography with the rest of ACBD3 being modelled by NMR and predictive modelling software (Figure 1). The Q domain is a glutamine rich region (aa241-308) which forms a long loop made of alpha helices [8]. The N-terminal ACB domain binds Acyl CoA and Palmitoyl CoA in other ACBD family proteins but the function of this domain in ACBD3 is unclear. To the N-terminus of the ACB domain is a proline rich region (aa21-60), which is indicative of protein-protein interaction sites and may complement the ACB domain whichis often found paired with protein-protein interaction domains such as the Pleckstrin homology domain (PH) and the Src homology domain in other proteins. ACBD3 makes essential interactions with an unusually high number of protein partners in cellular processes as diverse as Golgi structure, steroid synthesis and glucose import; other functions not reviewed here include iron transport and a causal role in Huntington’s disease progression [7,9-12]. ACBD3 is essential for neural development and human cell lines do not divide when ACBD3 is excised by CRISPR-CAS9 [13].
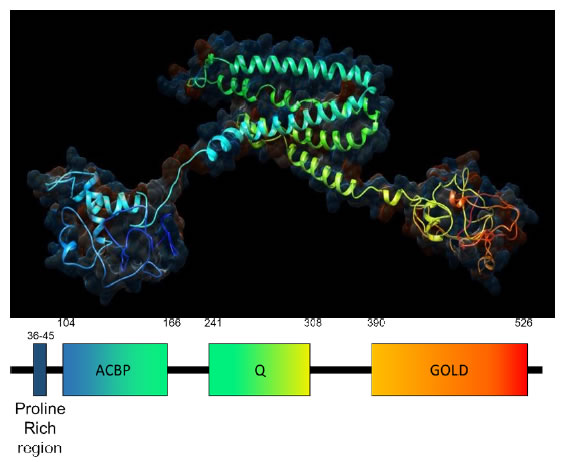
Figure 1. Predicted 3D structure of human ACBD3.The structure is modelled by Phyre2 software using the primary amino acid sequence which agrees strongly with crystal structures of individual ACBD3 domains and related proteins [86]. From the N-terminus in blue to the C-terminus in red ACBD3 clearly contains 3 domains: the ACBP domain, the Q domain and the Golgi dynamics (GOLD domain) respectively connected by flexible linkers with an N terminal proline rich region. The hydrophilic surface of acbd3 has been superimposed on ACBD3 showing the electrostatic charge of the protein model with red depicting negative charge and blue depicting positive charge.
ACBD3 in Breast Cancer
The q arm of Chromosome 1 contains many genes important in cancer progression or tumour suppression: NRAS, JUN, MYCL, ESRRG, ARF1 and RAB25are amongst the best known. There are however many more 1q genes that are amplified in breast cancer with deletions strikingly rare despite common deletions in the p arm. Some of these genes (PI4Kβ, PIP5K1A and HIST2H2BE) have more recently been recognised as oncogenic with ACBD3 being the latest 1q gene observed to affect breast cancer [14]. ACBD3 mRNA is reported to be upregulated in breast tumour tissue matched against adjacent normal tissue in all subtypes [5]. Protein levels of ACBD3 were upregulated in 8 breast cancer cell lines (MDA-MB453, MDA-MB-415, BT549, MDA-MB-231, ZR-75-30, SKBR3, T47D and MCF7) compared to 2 normal breast epithelial cell samples (NBEC1 and NBEC2). In a cohort of Chinese breast cancer patients ACBD3 protein expression increased as cancer stage became more advanced. Kaplan-Meier survival curves were plotted and it was found that high levels of ACBD3 mRNA in breast tumour tissue predicted lower rates of patient survival and that this made a large difference in stage III and IV cancers with 60% probability of survival at 120 months when ACBD3 expression is low but less than 30% probability of survival when ACBD3 expression is high.
The ACBD3 containing 1q42.12 locus is seen to be amplified in an additional 6 breast cell lines (BRCAMZ01, BT20, HCC2218, MDAMB436, SUM149, ZR751) and 6 out of 25 primary breast tumours in a breast cancer 1q amplification study [4]. Loss of region 1q42.12 was seen in only 1 cell line (UACC812) where the terminal ~38 megabases of arm 1q were deleted and loss of 1q42.12 was not observed in any primary tumour samples. RNA expression levels revealed that 1q42.12 is located in the middle of a region of gain coined G7, the largest region of gain (in bases) on chromosome 1 in breast cancers.Overexpression of ACBD3 in cell cultures caused increased side populations of stemlike cancer cells and inhibition of ACBD3 by siRNAsignificantly reduced these populations [5]. GSEA analysis found that CTNNB1- and TCF4-activated gene signatures both positively correlated with ACBD3 expression [5]. CTNNB1 encodes the β-catenin protein which, in response to Wnt signalling, accumulates in the cytoplasm and then translocates to the nucleus where it propagates the Wnt signal.ACBD3 overexpression led to an increase of β-catenin in the cytoplasm and nucleus compared to when ACBD3 expression was low (65% versus 20% nuclear and cytoplasmic localisation) [5]. TCF4 is a transcription factor for genes that code proteins in the Wnt signalling pathway. When TCF4 was knocked down, the self-renewal ability of ACBD3-expressing cells was abolished suggesting that ACBD3 may promote cancer stem cell propagation via the Wnt/ β-catenin signalling pathway in breast cancer.All of this provides strong evidence that ACBD3 overexpression affects breast cancer but the ACBD3 protein has many binding partners, in disparate cellular pathways and cells appear to have few redundancies for the essential roles of ACBD3.
ACBD3 and Steroidogenesis
Although often considered a resident Golgi protein due its structural role and interactions with other structural components, ACBD3 can also be found at other membranes including the cytosolic cell membrane and at the outer mitochondrial membrane (OMM). ACBD3 interacts withtranslocator protein TSPO (previously the peripheral-type benzodiazepine receptor) on the cytosolic OMM and stimulates cholesterol transport from the OMM to the IMM (inner mitochondrial membrane) (Figure 2) [7,15]. P450scc (CYP11A1) makes direct contact with the IMM and converts cholesterol to pregnenolone, the precursor to mammalian steroids, by side chain cleavage [16,17]. TSPO is anchored to the voltage dependent anion channel VDAC1 and makes up approximately 2% of OMM proteins. TSPO tethers cytosolic ACBD3 at the OMM and ACBD3 subsequently recruits protein kinase A (PKA) via the PKARIα subunit. This brings PKA into proximity with one of its substrates, the steroidogenic acute regulatory (StAR) protein which is phosphorylated on residues S57 and S195 by PKA [18]. StAR then facilitates cholesterol import from the OMM to the IMM, the rate limiting step in steroidogenesis. ACBD3 overexpression increases chorionic gonadotropin-induced steroid production; increased steroid production has obvious implications for cancer progression by enabling self-sufficiency in growth signals, a hallmark of cancer [19,20].
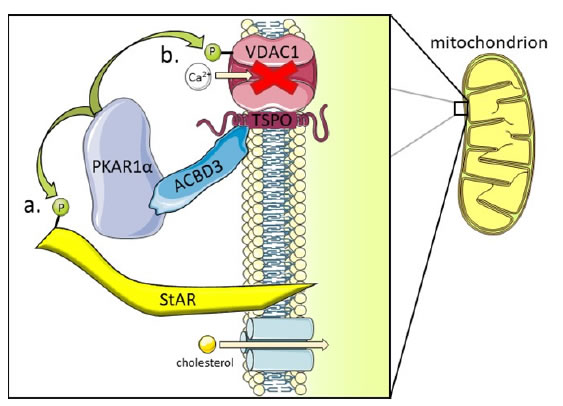
Figure 2. ACBD3 in redox stress and steroidogenesis. ACBD3 is a Golgi resident protein but also has functions elsewhere in the cell. At the Outer Mitochondrial membrane (OMM) ACBD3 is essential for mediating interactions between PKA holoenzyme (via direct tethering with the PKAR1α subunit shown) and two of its substrates: StAR and VDAC1.
a) The phosphorylation state of StAR determines whether cholesterol can cross the IMM and be converted to pregnenolone, the basic building block of all steroid hormones in mammals. Cholesterol import is the rate limiting step in steroidogenesis and ACBD3 is indispensable for this process [18].
b) VDAC1 is a Ca2+ ion import channel at the OMM and phosphorylation by PKA closes this channel to prevent calcium import. Mitochondrial import of Ca2+ forms part of the calcium homeostasis mechanism in the cell, closing the VDAC1 ion channel causes cytosolic Ca2+ concentration to rise in the cell which leads to redox stress and local inflammation. Again, ACBD3 is essential for localising PKA to the mitochondria where it can then phosphorylate the VDAC1 substrate [27].
PKAR1α is a tumour suppressor gene and is important in primary pigmented nodular adrenocortical disease (PPNAD) nodule formation and tumorigenesis in mice and humans. Mutation of PKAR1α leads to hypercortisolism that drives tumorigenesis, and high ACBD3 expression in steroidogenic tissues (of which the adrenal cortex is one) may contribute to the overexpression/over activity of the mutant PKAR1α [21]. PPNADs are characterised by a resistance to apoptosis which in itself contributes to cancer occurrence and is another hallmark of cancer [20]. The first publication to suggest any link between ACBD3 and cancer demonstrated that ACBD3 follows the same expression profile as PKAR1α in PPNAD tissue and speculated that, in tumorigenesis, this could lead to deregulation of steroid synthesis [21]. More recent studies have shown that PKA activation may instead have a suppressive effect on cancer [22,23], whilst others show PKARIα is upregulated in cancer cell lines [24,25].
ACBD3 in Redox Stress
In a separate process, glutamate induces expression of TSPOand increased TSPO recruits ACBD3 and PKA to the mitochondria. Glutamate is a signalling molecule that is known to cause acute neurotoxicity [26-28]. PKA phosphorylates the calcium channel protein VDAC1, preventing Ca2+ import into the mitochondria (Figure 2). This causes Ca2+ accumulation in the cytosol which signals redox stress via the calcium sensing CamKII and its effector NADPH oxidase (NOX5) leading to inflammation by increased reactive oxygen species (ROS). Glutamate mediated redox stress is particularly important in neuro-inflammationwhere TSPO is not expressed in healthy brain tissue but can accumulate in age related degenerative disease or after traumatic stress, leading to increased ACBD3 mitochondrial recruitment and subsequent VDAC1 phosphorylation by PKA which may contribute to neurodegeneration [29]. VDAC1 is important in Ca2+ homeostasis, especially mitochondrial Ca2+ homeostasis which controls the metabolism of mitochondria and therefore energy availability in the cell [30]. Dysregulation of cellular energetics is a hallmark of cancer and an inflammatory environment can be tumour promoting when chronic and over time [20].
ACBD3 and Insulin Mediated Glucose Import
GLUT4 (glucose transporter type 4) allows the facilitated diffusion of glucose from the surroundings into cells,andis sequestered into storage vesicles (GSVs) that are tethered to Golgi membranes by TUG (Tether containing UBX domain for GLUT4), Golgin-160 and ACBD3 when insulin is absent (Figure 3) [31]. ACBD3-bound TUG can be acetylated on lysine 549 which has a higher binding affinity with Golgin-160 than with ACBD3 [10]. In response to insulin receptor activation the cytoplasmic effector of insulin PIST (PDZ interacting specifically with TC10) binds Golgin-160 and catalyses the cleavage of acetylated TUG. This releases GSVs allowing them to fuse with the plasma membrane where GLUT4 forms a channel for glucose import [32]. GLUT4 is continuously cycled away from plasma membranes back into GSVs to increase the on/off response of insulin sensitive cells when insulin is not present. GLUT4 exocytosis is regulated by tankyrase 1 as are several other ACBD3 related processes including Golgin45 expression and the promotion of β-catenin transcription in the Wnt signalling pathway [33,34].
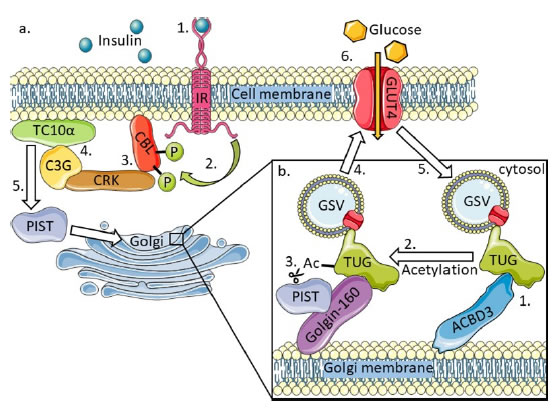
Figure 3. The effect of insulin on TUG, the interaction between TUG and ACBD3, and the recycling of GLUT4 storage vesicles to regulate glucose import [10,31].
a) 1. Extracellular insulin binds the transmembrane insulin receptor (IR) causing receptor activation. 2. The active IR tyrosine phosphorylates CBL inside the cell. 3. Phosphorylated CBL recruits the CRK-C3G complex to the membrane lipid raft sub domain facilitating interaction of C3G and TC10α. 4. C3G activates TC10α which subsequently activates its effector: PIST. 5. PIST relocates to the Golgi causing the release of GSVs with embedded GLUT4 transporter which fuse with the cell membrane, 6. allowing glucose to enter the cell. GLUT4 is continuously cycled away from the membrane in GSVs creating a fast on and off switch for insulin dependent glucose import.
b) 1. ACBD3 interacts with TUG and is dependent on the acetylation state of TUG. 2. Acetylation of TUG on lysine 549 causes TUG to preferentially bind Golgin-160 over ACBD3. 3. PIST, activated by the insulin receptor signalling cascade, also binds Golgin-160 and catalyses the cleavage of acetylated TUG causing GSVs to be released into the cytoplasm 4. to fuse with the cell membrane. 5. GLUT4 is continuously cycled away from the cell membrane embedded in GSVs and is sequestered back to the Golgi where they bind TUG.
The hormone 17β-oestradiolhas a central role in breast cancer progression, ithas been found to upregulateGLUT4 expression and translocation to the membranein breast cancer cell lines and was associated with increased glucose uptake [35-37]. GLUT4 is being investigated as a target for breast cancer therapy as part of an informed approach to target the Warburg effect. Downregulation of GLUT4 by siRNAimpairs viability of MDA-MB-231 and MCF7 breast cancer cell lines and increases mitochondrial oxidation of pyruvate [38]. The EGFR/HER2-targeted drug lapatinib has been shown to downregulate GLUT4 in ER-/HER2+ HMEC cell lines, and GLUT4 downregulation by siRNA in these cell lines led to the formation of normal acini structures in 3D culture [39].The insulin receptor (IR) is upregulated in breast cancer and is a potential target for breast cancer therapy as it has been demonstrated that knock down of IR by shRNA and inhibition by peptide drugs inhibits breast cancer cell growth [40-43].
ACBD3, PI4Kβand Phosphatidylinositols
Phosphatidylinositol 4 Kinase III beta (PI4Kβ) is a lipid kinase that converts phosphatidylinositol (PI) into phosphatidylinositol 4-phosphate (PI(4)P) [44]. PI4Kβ is localised to the Golgi by extension of an amphipathic helix at the N-terminus of PI4Kβ (aa44-64) through the Q domain alpha helices loop of ACBD3 (aa241-308) [8]. The small GTPase Rab11 binds PI4Kβ to support this interaction whilst ACBD3 also interacts with GOLGB1 on the Golgi surface bringing PI4Kβ in close and constant contact withits PI substrate embedded in the lipid bilayer. ACBD3 does not affect the enzymatic activity of PI4Kβ directly by this interaction but, by tethering it to the Golgi membrane, PI4Kβ is proximal to the PI substrate and does not rely on diffusion through the cytoplasm for the phosphorylation of substrate. PI4Kβ is heavily implicated in breast cancers with 20% of primary tumours showing over expression of PI4Kβ at the protein level [45,46]. PI4Kβ is a chromosome 1q gene (at 1q21.3) and is reported to have increased gene copy number in 62% of 939 patient breast tumour samples [14]. Evidence of PI4Kβ upregulation at the protein level in breast ductal carcinoma samples from the human protein atlas was also found by Waugh. Independent of its lipid kinase function, PI4KIIIβ also mediates indirect phosphorylation and activation of AKT (Protein kinase B), an important kinase in breast cancer signalling [46,47]. AKT dysregulation drives many breast cancers by promoting cell cycle progression and suppressing apoptosis, it is commonly overexpressed or constitutively active [47].
Both PI and PI(4)P are cellular signalling molecules and docking sites on the membrane for other proteins including ARF1 (ADP-ribosylation Factor 1), which is essential for the formation of COPI vesicles and Golgi function including localisation of Golgin-160 to the Golgi; ARF1 is encoded by a gene adjacent to ACBD3 on chromosome 1 (1q42.13) [48-50]. PI4Kβ is also positioned on chromosome 1q, where amplification is common in breast cancers and its substrates localise ARF1 to membranes. ACBD3 is hijacked by some picornavirusviral proteins to form replication organelles and recruits PI4Kβ to these sites to enrich them for PI(4)P [51,52]. This is another example of how the role of ACBD3 is contextual and dependent on its cellular location, cell cycle position and binding partners. PI4Kβ has been found to be a target in malaria and drugs to inhibit PI4Kβ have already been developed [53]. There have so far been no publications on PI4Kβ drug inhibition in cancer.
ACBD3 Cell Signalling in Neurogenesis
Mammalian NUMB, an endocytic adapter protein, is involved in cytosolic signalling and is segregated asymmetrically into one daughter cell during the mitosis of neural progenitor cells and inhibits NOTCH [54]. This asymmetric distribution of NUMB results in 1 identical pluripotent daughter cell (high NUMB protein level) to maintain the population of neuronal precursors and 1 differentiated neuron cell (low NUMB protein level). This balances the need to create neurons through NOTCH signalling and maintain the pool of precursor cells in embryonic neurogenesis by NOTCH inhibition (Figure 4) [55,56]. ACBD3 was identified as a NUMB binding partner after observations that ACBD3 cytosolic release during mitosis was paired with NUMB mediated cell fate [57]. The ACBD3 interacting region on NUMB is essential for NUMB activity and interaction with ACBD3 increases NUMB activity [57]. The C-terminus of ACBD3 binds with the N-terminus of NUMB, and NOTCH also binds the N-terminus of NUMB [58]. Cytosolic ACBD3 expression leads to inhibition of NOTCH, suggesting that NOTCH inhibition by NUMB is conserved from drosophila to mammals indicating that ACBD3 and NUMB are both required to specify cell fate in neural progenitors. ACBD3 is bound to Golgi/mitochondrial membranes through most of the cell cycle and can only bind NUMB during mitosis when the breakdown of the Golgi releases ACBD3 into the cytosol (Figure 4). Constitutively cytosolic mutant ACBD3 inhibits neurogenesis in mouse embryos resulting in fewer neurons. This indicates that permanently cytosolic ACBD3 is preventing differentiation in otherwise neuronal fated cells and it achieves this by binding NUMB outside of mitosis [57].
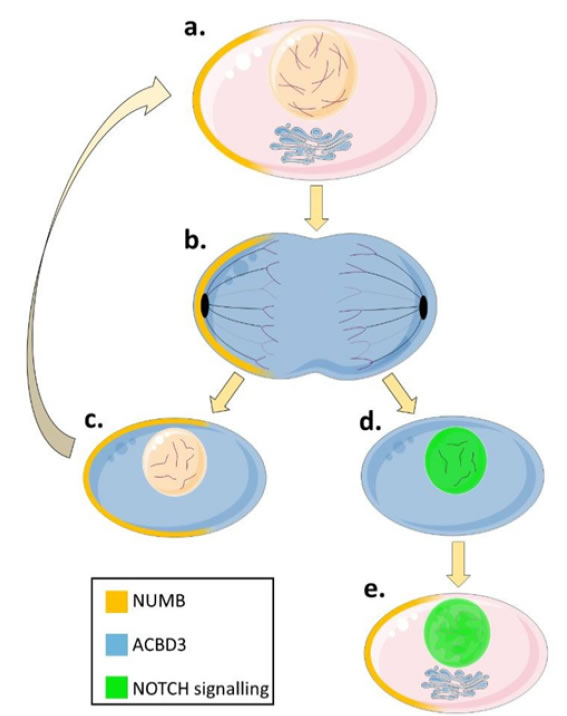
Figure 4. The differential regulation of NOTCH signalling by ACBD3 and NUMB in neurogenesis. Cytosolic NUMB (yellow shading) acts synergistically with cytosolic ACBD3 (blue shading) to inhibit NOTCH signalling (represented by a green nucleus) and specifies progenitor cell fates during mitosis [57]. In one daughter cell, cytosolic ACBD3 binds to NUMB increasing the ability of NUMB to inhibit NOTCH and causing that daughter cell to remain a progenitor like its parent. The other daughter cell does not contain NUMB and so NOTCH signalling cannot be prevented despite the presence of cytosolic ACBD3 protein and so the daughter cell begins to differentiate into a neuron. Paradoxically NUMB quickly accumulates in the differentiating cell but because the Golgi has reformed at this point, ACBD3 is membrane bound and cannot influence NUMB-NOTCH protein interactions, instead NUMB promotes neuron differentiation and survival through other pathways. This leads to the creation of one daughter neuron and one progenitor cell to balance neurodevelopment and stem cell pools.
a) NUMB accumulates asymmetrically in one half of a progenitor cell before mitosis. ACBD3 is bound to the Golgi apparatus and other organelles (not shown) and does not interact with NUMB.
b) During mitosis, the Golgi apparatus fragments into vesicles and ACBD3 is released into the cytosol where it can interact with asymmetrically distributed NUMB.
c) One of the daughter cells will contain NUMB and cytosolic ACBD3. ACBD3 increases the ability of NUMB to bind and inhibit NOTCH. Without NOTCH signalling the daughter cell remains a progenitor cell to maintain the pool of neuronal precursors.
d) The second daughter cell will contain cytosolic ACBD3 but no NUMB protein meaning that NOTCH is not inhibited and enters the nucleus signalling to the cell to differentiate.
e) NUMB protein is produced in the differentiating cell in G1 but at this time the Golgi has reformed and ACBD3 is no longer free in the cytoplasm and cannot interact with NUMB and so cannot inhibit NOTCH signalling.
Krüppel-like factor 9 (KLF9) is a tumour suppressor and is significantly down regulated in invasive breast cancers, endometrial carcinoma, glioblastoma and colorectal cancer, and its expression can inhibit growth of tumour xenografts from glioblastomaneurospheres [59,60]. ACBD3 and NOTCH1 expression are suppressed by KLF9 in endometrial carcinoma cells and both proteins promote breast cancer progression, specifically in cancer stem cell maintenance [5,61,62]. KLF9 supresses glioblastomaderivedneurosphere formation by 60% in controls but only by 33% when NOTCH1 expression is constitutively active strongly suggesting that KLF9 must suppress other proteins relevant to glioblastoma cancer progression and this could include ACBD3 [59]. NOTCH1, NOTCH3 and JAG1expression isassociated with poor survival in breast cancer patients with high NOTCH1 expression conferring a 66% chance of mortality and 74% chance of relapse at 10 years [63]. NOTCH receptorshave been found to have activating mutations in triple negative breast cancers and result in the upregulation of NOTCH controlled genes [64]. NOTCH overexpression was able to transform the MCF10A breast cell line and reduce its sensitivity to apoptotic drugs such as staurosporine, melphalan, or mitoxantrone, and overexpression of NUMB reverted the transformation [61].High levels of NOTCH in breast tumours are significantly associated with nuclear phospho-Erk 1 and 2 conferring an association between NOTCH and Ras-MAPK expression [65]. Inhibition of NOTCH and NOTCH-related proteinshave therefore become a target for therapy [66]. ACBD3 overexpression prevents NOTCH signalling in neurogenesis but this is reliant on NUMB expression and only during mitosis when the Golgi is fragmented. The NOTCH suppressorsNUMB and its paralogue NUMB-L are predictably down regulated in breast cancers and their overexpression reduces epithelial to mesenchymal transition in triple negative breast cancer cell lines [67-69]. NUMB-deficient breast cancer cells have an increased ability to form cancer stem cell pools and NUMBdownregulation causes inactivation of p53 [70,71].
Discussionand Future Perspectives
Throughout this review an argument is presented that ACBD3 may do more than promote Wnt signalling in the context of breast cancer. Dysregulation of cellular energetics, sustaining proliferative signalling, replicative immortality andtumour-promoting inflammation are all hallmarks of cancer and overexpression of ACBD3 could conceivably support any or all of these [10,18,21,27,57]. Other factors including the position of ACBD3 on chromosome 1 in close proximity to other oncogenes, and the number of its binding partners and pathways already being targeted for cancer therapies leave ACBD3 nothing short of overlooked.ARF1 and RAB4 are located close to ACBD3, both at 1q42.13, withinOrsetti’s(4) region of gain G6 and were both found to be significantly overexpressed at the mRNA level in breast cancer. RAB4 in conjunction with RAB5 promotes and drives metastasis by facilitating the formation of invadosomes containing membrane type 1 matrix metalloprotease (MT1-MMP) and β3 integrin which together degrade the extracellular matrix, a process vital for cancer invasion and metastasis [72]. RAB4 is overexpressed in breast cancers and unsurprisingly associated with increased cell motility, it is one of many RAS related proteins that has clinical significance in cancer [73]. ARF1 is the most amplified gene of the ADP-ribosylation factor family in breast cancers and its amplification is associated with increased gene transcription and worse prognosis for patients [74]. ARF1 inhibition prevents metastasis of tumour xenografts in immuno-deficient mice and is replicable in zebrafish models of breast cancer metastasis. ACBD3 proximity to ARF1, RAB4 and other 1q oncogenes may confer a huge selective advantage to breast cancer cells with amplifications of these lociproviding these cells with both survival and invasive advantages.
Tankyrase 1 regulates GLUT4 exocytosis and β-catenintranscription,and ACBD3 interacts with proteins in both pathways [75-78]. Tankyrase 1 controls the expression of Golgin45 which is a direct binding partner of ACBD3 [79]. Tankyrase also targets Axin for degradation leading to increased Wnt signalling, known to be aberrant in breast cancers andis reported to be effected by ACBD3 [5,76,80]. Tankyrase 1 and 2 are currently being targeted as cancer therapeutics because of their interactions inmany carcinogenic pathways [77,81-83]. PI4Kβ expression in breast cancer correlates with poor patient outcomes and its locus (1q21.3) is a biomarker for breast cancer [46,84]. It is most associatedas an ACBD3 binding partner and the ACBD3 interaction has a solved X-ray crystal structure [8]. PI4Kβ mutants that do not bind ACBD3 have been engineered and drugs that inhibit PI4Kβ are available which aids its study [53,85]. ACBD3deletion is embryonic lethal and may be invaluable for normal cell division [12,57]. As it does not have an enzymatic function an inhibitor for ACBD3 may not be viable and non-targeteddownregulation may not be desirable, instead targeting one or more of its protein-protein interactions or partners may be the route to new treatments in breast cancer.
References
- Fridlyand J, Snijders AM, Ylstra B, Li H, Olshen A, et al. (2006) Breast tumor copy number aberration phenotypes and genomic instability. BMC Cancer 6: 96. [Crossref]
- Tirkkonen M, Tanner M, Karhu R, Kallioniemi A, Isola J, et al. (1998) Molecular cytogenetics of primary breast cancer by CGH. Genes Chromosomes Cancer 21: 177-184. [Crossref]
- Soloviev M, Esteves MP, Amiri F, Crompton MR, Rider CC (2013) Elevated transcription of the gene QSOX1 encoding quiescin Q6 sulfhydryl oxidase 1 in breast cancer. PloSOne8: e57327. [Crossref]
- Orsetti B, Nugoli M, Cervera N, Lasorsa L, Chuchana P, et al. (2006) Genetic profiling of chromosome 1 in breast cancer: mapping of regions of gains and losses and identification of candidate genes on 1q. Br J Cancer 95: 1439-1447. [Crossref]
- Huang Y, Yang L, Pei Y, Wang J, Wu H, et al. (2018) Overexpressed ACBD3 has prognostic value in human breast cancer and promotes the self-renewal potential of breast cancer cells by activating the Wnt/beta-catenin signaling pathway. Exp Cell Res 363:39-47. [Crossref]
- Sohda M, Misumi Y, Yamamoto A, Yano A, Nakamura N, et al. (2001) Identification and Characterization of a Novel Golgi Protein, GCP60, That Interacts with the Integral Membrane Protein Giantin. J BiolChem 276:45298-45306. [Crossref]
- Li H, Degenhardt B, Tobin D, Yao Z, Tasken K, et al. (2001) Identification, Localization, and Function in Steroidogenesis of PAP7: A Peripheral-Type Benzodiazepine Receptor- and PKA (RIalpha)-Associated Protein. MolEndocrinol 15:2211-2228. [Crossref]
- Klima M, Tóth DJ, Hexnerova R, Baumlova A, Chalupska D, et al. (2016) Structural insights and in vitro reconstitution of membrane targeting and activation of human PI4KB by the ACBD3 protein. Scientific Reports6:23641. [Crossref]
- Xihua Y, Mengjing B, Romain C, Siyang L, Jia M, et al. (2017) ACBD3 functions as a scaffold to organize the Golgi stacking proteins and a Rab33b-GAP. FEBS Lett 591:2793-802. [Crossref]
- Belman JP, Bian RR, Habtemichael EN, Li DT, Jurczak MJ, et al. (2015) Acetylation of TUG Protein Promotes the Accumulation of GLUT4 Glucose Transporters in an Insulin-responsive Intracellular Compartment. J BiolChem 290:4447-4463. [Crossref]
- Sbodio JI, Paul BD, Machamer CE, Snyder SH (2013) Golgi protein ACBD3 mediates neurotoxicity associated with Huntington’s disease. Cell reports 4:890-897. [Crossref]
- Okazaki Y, Ma Y, Yeh M, Yin H, Li Z, et al. (2012) DMT1 (IRE) expression in intestinal and erythroid cells is regulated by peripheral benzodiazepine receptor-associated protein 7. Am J PhysiolGastrointest Liver Physiol 302:G1180-1190. [Crossref]
- Lyoo H, van der Schaar, Hilde M, Dorobantu CM, Rabouw HH, et al. (2019) ACBD3 Is an Essential Pan-enterovirus Host Factor That Mediates the Interaction between Viral 3A Protein and Cellular Protein PI4KB. mBio 10:e02742-18. [Crossref]
- Waugh MG (2014) Amplification of Chromosome 1q Genes Encoding the Phosphoinositide Signalling Enzymes PI4KB. AKT3, PIP5K1A and PI3KC2B in Breast Cancer. J Cancer 5:790-796. [Crossref]
- Krueger KE, Papadopoulos V (1990) Peripheral-type benzodiazepine receptors mediate translocation of cholesterol from outer to inner mitochondrial membranes in adrenocortical cells. J BiolChem 265:15015-15022. [Crossref]
- Strushkevich N, MacKenzie F, Cherkesova T, Grabovec I, Usanov S, et al. (2011) Structural basis for pregnenolone biosynthesis by the mitochondrial monooxygenase system. ProcNatlAcadSci U S A 108:10139-10143. [Crossref]
- Elustondo P, Martin LA, Karten B (2017) Mitochondrial cholesterol import. BiochimBiophysActaMol Cell Biol Lipids 1862:90-101. [Crossref]
- Arakane F, King SR, Du Y, Kallen CB, Walsh LP, et al. (1997) Phosphorylation of Steroidogenic Acute Regulatory Protein (StAR) Modulates Its Steroidogenic Activity. J BiolChem 272:32656-32662. [Crossref]
- Hanahan D, Weingberg R (2000) The hallmarks of cancer. Cell 100:57-70. [Crossref]
- Hanahan D, Weinberg RA (2011) Hallmarks of Cancer: The Next Generation. Cell 144:646-674. [Crossref]
- Liu J, Matyakhina L, Han Z, Sandrini F, Bei T, et al. (2003) Molecular cloning, chromosomal localization of human peripheral-type benzodiazepine receptor and PKA regulatory subunit type 1A (PRKAR1A)-associated protein PAP7, and studies in PRKAR1A mutant cells and tissues. FASEB J 17:1189-1191. [Crossref]
- Pattabiraman DR, BierieB, Kober KI, Thiru P, Krall JA, et al. (2016) Activation of PKA leads to mesenchymal-to-epithelial transition and loss of tumor-initiating ability. Science 351:aad3680. [Crossref]
- Persaud L, Mighty J, Zhong X, Francis A, Mendez M, et al. (2018) IL-24 Promotes Apoptosis through cAMP-Dependent PKA Pathways in Human Breast Cancer Cells. Int J MolSci 19:3561. [Crossref]
- Mantovani G, Bondioni S, Lania AG, Rodolfo M, Peverelli E, et al. (2008) High expression of PKA regulatory subunit 1A protein is related to proliferation of human melanoma cells. Oncogene 27:1834-1843. [Crossref]
- McDaid HM, Cairns MT, Atkinson RJ, McAleer S, Harkin DP, et al. (1999) Increased expression of the RIalpha subunit of the cAMP-dependent protein kinase A is associated with advanced stage ovarian cancer. Br J Cancer 79:933-939. [Crossref]
- Loilome W, Juntana S, Namwat N, Bhudhisawasdi V, Puapairoj A, et al. (2011) PRKAR1A is overexpressed and represents a possible therapeutic target in human cholangiocarcinoma. Int J Cancer 129:34-44. [Crossref]
- Gatliff J, East DA, Singh A, Alvarez MS, Frison M, et al. (2017) A role for TSPO in mitochondrial Ca2+ homeostasis and redox stress signaling. Cell Death Dis8:e2896. [Crossref]
- Atlante A, Calissano P, Bobba A, Giannattasio S, Marra E, (2001) Glutamate neurotoxicity, oxidative stress and mitochondria. FEBS Lett 497:1-5. [Crossref]
- Kumar A, Muzik O, Shandal V, Chugani D, Chakraborty P, et al. (2012) Evaluation of age-related changes in translocator protein (TSPO) in human brain using (11)C-[R]-PK11195 PET. J Neuroinflammation 9:232. [Crossref]
- Shoshan-Barmatz V, Krelin Y, Shteinfer-Kuzmine A (2018) VDAC1 functions in Ca2+ homeostasis and cell life and death in health and disease. Cell Calcium69:81-100. [Crossref]
- Bogan JS, Rubin BR, Yu C, Löffler MG, Orme CM, et al. (2012) Endoproteolytic Cleavage of TUG Protein Regulates GLUT4 Glucose Transporter Translocation. J BiolChem 287:23932-23947. [Crossref]
- Mohan S, Sheena A, Poulose N, Anilkumar G (2010) Molecular Dynamics Simulation Studies of GLUT4: Substrate-Free and Substrate-Induced Dynamics and ATP-Mediated Glucose Transport Inhibition. PLoS One 5:e14217. [Crossref]
- Guo H, Zhang C, Liu Q, Li Q, Lian G, et al. (2012) The Axin/TNKS complex interacts with KIF3A and is required for insulin-stimulated GLUT4 translocation. Cell Res22:1246-1257. [Crossref]
- Huang SA, Mishina YM, Liu S, Cheung A, Stegmeier F, et al. (2009) Tankyrase inhibition stabilizes axin and antagonizes Wnt signalling. Nature 461:614-620. [Crossref]
- Russo J, Russo IH (2006) The role of estrogen in the initiation of breast cancer. J Steroid BiochemMolBiol 102:89-96. [Crossref]
- Miller WR, O’Neill J (1987) The importance of local synthesis of estrogen within the breast. Steroids 50:537-548. [Crossref]
- Neeman M, Degani H (1989) Metabolic Studies of Estrogen- and Tamoxifen-treated Human Breast Cancer Cells by Nuclear Magnetic Resonance Spectroscopy. Cancer Res 49:589. [Crossref]
- Garrido P, Osorio FG, Morán J, Cabello E, Alonso A, et al. (2015) Loss of GLUT4 induces metabolic reprogramming and impairs viability of breast cancer cells. J Cell Physiol 230:191-198. [Crossref]
- Acharya S, Xu J, Wang X, Jain S, Wang H, et al. (2016) Downregulation of GLUT4 contributes to effective intervention of estrogen receptor-negative/HER2-overexpressing early stage breast disease progression by lapatinib. Am J Cancer Res 6:981-995. [Crossref]
- Chan JY, LaPara K, Yee D (2016) Disruption of insulin receptor function inhibits proliferation in endocrine-resistant breast cancer cells. Oncogene 35:4235-4243. [Crossref]
- Rostoker R, Abelson S, Bitton-Worms K, Genkin I, Ben-Shmuel S, et al. (2015) Highly specific role of the insulin receptor in breast cancer progression. EndocrRelat Cancer 22:145-157. [Crossref]
- Papa V, Pezzino V, Costantino A, Belfiore A, Giuffrida D, et al. (1990) Elevated insulin receptor content in human breast cancer. J Clin Invest 86:1503-1510. [Crossref]
- Chan JY, Hackel BJ, Yee D (2017) Targeting Insulin Receptor in Breast Cancer Using Small Engineered Protein Scaffolds. Mol Cancer Ther 16:1324. [Crossref]
- Meyers R, Cantley LC (1997) Cloning and characterization of a wortmannin-sensitive human phosphatidylinositol 4-kinase. J BiolChem 272:4384-4390. [Crossref]
- Tan J, Brill JA (2014) Cinderella story: PI4P goes from precursor to key signaling molecule. Crit Rev BiochemMolBiol 49:33-58. [Crossref]
- Morrow AA, Alipour MA, Bridges D, Yao Z, Saltiel AR, et al. (2014) The Lipid Kinase PI4KIIIβ Is Highly Expressed in Breast Tumors and Activates Akt in Cooperation with Rab11a. Mol Cancer Res 12:1492. [Crossref]
- Paplomata E, O’Regan R (2014) The PI3K/AKT/mTOR pathway in breast cancer: targets, trials and biomarkers. TherAdv Med Oncol 6:154-166. [Crossref]
- Yadav S, Puthenveedu M, Linstedt A (2012) Golgin160 Recruits the Dynein Motor to Position the Golgi Apparatus. Dev Cell 23:153-165. [Crossref]
- Lee MCS, Miller EA, Goldberg J, Orci L, Schekman R (2004) Bi-directional Protein Transport Between the ER and Golgi. Annu Rev Cell DevBiol 20:87-123. [Crossref]
- Liu Y, Kahn RA, Prestegard JH (2014) Interaction of Fapp1 with Arf1 and PI4P at a membrane surface: an example of coincidence detection. Structure 22:421-430. [Crossref]
- Sasaki J, Ishikawa K, Arita M, Taniguchi K (2012) ACBD3-mediated recruitment of PI4KB to picornavirus RNA replication sites. EMBO J 31:754. [Crossref]
- Xiao X, Lei X, Zhang Z, Ma Y, Qi J, et al. (2017) Enterovirus 3A facilitates viral replication by promoting PI4KB-ACBD3 interaction. J Virole00791-17. [Crossref]
- McNamara CW, Lee MC, Lim CS, Lim SH, Roland J, et al. (2013) Targeting Plasmodium PI(4)K to eliminate malaria. Nature 504:248-253. [Crossref]
- Artavanis-Tsakonas S, Rand MD, Lake RJ (1999) Notch Signaling: Cell Fate Control and Signal Integration in Development. Science 284:770. [Crossref]
- Uemura T, Shepherd S, Ackerman L, Jan LY, Jan YN (1989) numb, a gene required in determination of cell fate during sensory organ formation in Drosophila embryos. Cell 58:349-360. [Crossref]
- Verdi JM, Schmandt R, Bashirullah A, Jacob S, Salvino R, et al. (1996) Mammalian NUMB is an evolutionarily conserved signaling adapter protein that specifies cell fate. Current Biology 6:1134-1145. [Crossref]
- Zhou Y, Atkins JB, Rompani SB, Bancescu DL, Petersen PH, et al. (2007) The Mammalian Golgi Regulates Numb Signaling in Asymmetric Cell Division by Releasing ACBD3 during Mitosis. Cell 129:163-178. [Crossref]
- Guo M, Jan LY, Jan YN (1996) Control of Daughter Cell Fates during Asymmetric Division: Interaction of Numb and Notch. Neuron 17:27-41. [Crossref]
- Ying M, Tilghman J, Wei Y, Guerrero-Cazares H, Quinones-Hinojosa A, et al. (2014) Kruppel-like Factor-9 (KLF9) Inhibits GlioblastomaStemness through Global Transcription Repression and Integrin α6 Inhibition. J BiolChem 289:32742-32756. [Crossref]
- Limame R, de Beeck KO, Van Laere S, Croes L, De Wilde A, et al. (2014) Expression profiling of migrated and invaded breast cancer cells predicts early metastatic relapse and reveals Krüppel-like factor 9 as a potential suppressor of invasive growth in breast cancer. Oncoscience 1:69-81. [Crossref]
- Stylianou S, Clarke RB, Brennan K (2006) Aberrant Activation of Notch Signaling in Human Breast Cancer. Cancer Res 66:1517-1525. [Crossref]
- Simmen FA, Su Y, Xiao R, Zeng Z, Simmen RCM (2008) The Krüppel-like factor 9 (KLF9) network in HEC-1-A endometrial carcinoma cells suggests the carcinogenic potential of dys-regulated KLF9 expression. ReprodBiolEndocrinol6:41. [Crossref]
- Reedijk M, Odorcic S, Chang L, Zhang H, Miller N, et al. (2005) High-level coexpression of JAG1 and NOTCH1 is observed in human breast cancer and is associated with poor overall survival. Cancer Res 65:8530-8537. [Crossref]
- Wang K, Zhang Q, Li D, Ching K, Zhang C, et al. (2015) PEST domain mutations in Notch receptors comprise an oncogenic driver segment in triple-negative breast cancer sensitive to a γ- secretase inhibitor. Clin Cancer Res 21:1487-1496. [Crossref]
- Mittal S, Subramanyam D, Dey D, Kumar RV, Rangarajan A (2009) Cooperation of Notch and Ras/MAPK signaling pathways in human breast carcinogenesis. Mol Cancer8:128,4598-8-128. [Crossref]
- Kontomanolis EN, Kalagasidou S, Pouliliou S, Anthoulaki X, Georgiou N, et al. (2018) The Notch Pathway in Breast Cancer Progression. ScientificWorldJournal2018:2415489. [Crossref]
- Zhang J, Shao X, Sun H, Liu K, Ding Z, et al. (2016) NUMB negatively regulates the epithelial-mesenchymal transition of triple-negative breast cancer by antagonizing Notch signaling. Oncotarget 7:61036-61053. [Crossref]
- García -Heredia JM, VerdugoSivianes EM, Lucena-Cacace A, Molina-Pinelo S, Carnero A (2016) Numb-like (NumbL) downregulation increases tumorigenicity, cancer stem cell-like properties and resistance to chemotherapy. Oncotarget 7:63611-63628. [Crossref]
- Kuchenbaecker KB, Hopper JL, Barnes DR, Phillips K, Mooij TM, et al. (2017) Risks of Breast, Ovarian, and Contralateral Breast Cancer for BRCA1 and BRCA2 Mutation Carriers. JAMA 317:2402-2416. [Crossref]
- Tosoni D, Pambianco S, EkalleSoppo B, Zecchini S, Bertalot G, et al. (2017) Pre-clinical validation of a selective anti-cancer stem cell therapy for Numb-deficient human breast cancers. EMBO Mol Med 9:655-671. [Crossref]
- Tosoni D, Zecchini S, Coazzoli M, Colaluca I, Mazzarol G, et al. (2015) The Numb/p53 circuitry couples replicative self-renewal and tumor suppression in mammary epithelial cells. J Cell Biol 211:845-862. [Crossref]
- Frittoli E, Palamidessi A, Marighetti P, Confalonieri S, Bianchi F, et al. (2014) A RAB5/RAB4 recycling circuitry induces a proteolytic invasive program and promotes tumor dissemination. J Cell Biol 206:307-328. [Crossref]
- Tzeng H, Wang Y (2016) Rab-mediated vesicle trafficking in cancer. J Biomed Sci23:70. [Crossref]
- Xie X, Tang S, Cai Y, Pi W, Deng L, et al. (2016) Suppression of breast cancer metastasis through the inactivation of ADP-ribosylation factor 1. Oncotarget 7:58111-58120. [Crossref]
- Su Z, Deshpande V, James DE, Stöckli J (2018) Tankyrase modulates insulin sensitivity in skeletal muscle cells by regulating the stability of GLUT4 vesicle proteins. J BiolChem 293:8578-8587. [Crossref]
- Zhang Y, Liu S, Mickanin C, Feng Y, Charlat O, et al. (2011) RNF146 is a poly(ADP-ribose)-directed E3 ligase that regulates axin degradation and Wnt signalling. Nat Cell Biol13:623. [Crossref]
- Kim MK (2018) Novel insight into the function of tankyrase. Oncol Lett 16:6895-6902. [Crossref]
- Kang DH, Lee DJ, Lee S, Lee S, Jun Y, et al. (2017) Interaction of tankyrase and peroxiredoxin II is indispensable for the survival of colorectal cancer cells. Nature Communications 8:40. [Crossref]
- Zhao J, Li B, Huang X, Morelli X, Shi N (2017) Structural Basis for the Interaction between Golgi Reassembly-stacking Protein GRASP55 and Golgin45. The J BiolChem 292:2956-2965. [Crossref]
- Howe LR, Brown AM (2004) Wntsignaling and breast cancer. Cancer Biol&Ther 3:36-41. [Crossref]
- Lu H, Lei Z, Lu Z, Lu Q, Lu C, et al. (2013) Silencing tankyrase and telomerase promotes A549 human lung adenocarcinoma cell apoptosis and inhibits proliferation. Oncol Rep 30:1745-1752. [Crossref]
- Seimiya H, Muramatsu Y, Ohishi T, Tsuruo T (2005) Tankyrase 1 as a target for telomere-directed molecular cancer therapeutics. Cancer Cell 7:25-37. [Crossref]
- Haikarainen T, Krauss S, Lehtio L (2014) Tankyrases: structure, function and therapeutic implications in cancer. Curr Pharm Des 20:6472-6488. [Crossref]
- Goh JY, Feng M, Wang W, Oguz G, Yatim SMJM, et al. (2017) Chromosome 1q21.3 amplification is a trackable biomarker and actionable target for breast cancer recurrence. Nat Med23:1319. [Crossref]
- Greninger AL, Knudsen GM, Betegon M, Burlingame AL, DeRisi JL (2013) ACBD3 interaction with TBC1 domain 22 protein is differentially affected by enteroviral and kobuviral 3A protein binding. mBio 4:e00098-13. [Crossref]
- Kelley LA, Mezulis S, Yates CM, Wass MN, Sternberg MJE (2015) The Phyre2 web portal for protein modeling, prediction and analysis. Nature Protocols10:845. [Crossref]